【复材资讯】最新AFM揭示O3型NaxFeyMn1−yO2正极材料中相演变、铁迁移与氧氧化还原的相互作用


【研究背景】
自20世纪90年代初商业化以来,可充电锂离子电池不可或缺,但其依赖稀缺有毒的镍、钴、锂。钠离子电池是更可持续的替代方案,其层状钠过渡金属(TM)氧化物正极材料因成本低、化学多样而备受关注,主要有P2型和O3型。由铁、锰等丰产元素组成的材料尤其有吸引力,如O3-NaxFeyMn1₋yO2容量超160 mAh/g,但常发生复杂结构转变、电压衰减和容量损失,后者与高电位下铁离子迁移和氧氧化还原有关。O3型相初始钠含量高、理论容量大,但循环中复杂相变导致堆叠无序和性能退化。我们近期对等摩尔比O3-Na0.95Fe0.5Mn0.5O2的研究揭示了充放电结构转变机制:首次循环经历O3→P3→OP2-1(充电)和OP2-1→OP2-2→O3(放电),后续循环演变为O3↔X可逆转变(图1)。无序OP2和X相由堆垛层错形成,含O型和P型结构域;OP2中O/P层交替形成小结构域,X相中O型结构域占75%以上。相变路径变化由氧化还原电位偏移主导:第二次循环时Fe3+_→Fe4+向高电位偏移,促使Fe3+迁移至O型NaO₂层中的四面体位置,锚定O型层,阻碍O3→P3相变,最终形成X相。但Fe浓度为0.5时迁移程度轻,仅能通过间接现象观测。因此,Fe迁移程度、发生电位、是否与氧氧化还原直接相关及可逆性仍是重要问题。富含铁的O3-NaxFeyMn1₋yO2(y≥0.5)鲜有探索(y>0.66报道极少)。高铁含量加剧结构不稳定和性能退化,但其结构根源尚不清楚。鉴于铁迁移和氧氧化还原在所有含铁O3型材料中起核心作用,高铁组成可放大铁氧化还原贡献、增强铁占据四面体的趋势,是研究铁迁移、氧活性与相稳定性相互作用的理想模型体系。
【工作简介】
在开发可持续钠离子电池正极材料时,利用铁等丰产元素对降低成本、减少稀缺过渡金属依赖至关重要。但含铁层状氧化物常伴有复杂的氧化还原行为与结构不稳定性,因此理解铁如何影响氧化还原特性及相变演化尤为关键。丹麦奥胡斯大学DortheB.Ravnsbæk以富铁O3型层状Na1₋yFexO2(y =0.5–0.8)为模型体系,结合循环伏安法、原位X射线吸收光谱及对堆垛层错敏感的原位粉末X射线衍射,阐明了铁的作用机制。结果表明,增加铁含量使高电位氧化还原反应向更低电位偏移,并促进充电过程中Fe3+更早迁移至O型NaO₂层的四面体位点。该迁移显著影响相变:中等铁含量样品在首次循环中可完成O3→P3→OP2→O3完整相变序列;而富铁材料因O型层早期发生结构“钉扎”,始终局限于具有堆垛层错的O3类结构。本研究阐明了铁在阳离子与阴离子氧化还原相互作用中的机理作用,揭示了为何铁含量对O3型钠离子正极结构稳定性的影响强烈且依赖于组成。相关成果以“Interplay Between Phase Evolution, Fe Migration, and Oxygen Redox in O3 Type NaxFeyMn1−yO2 Cathodes”为题发表在《Advanced Functional Materials》上。
【研究内容】
为理解O3-NNaxFeyMn1−yO2的氧化还原行为,结合循环伏安法(CV,图1A–C)及Fe/Mn K边XANES(图2)。因材料充放电中发生多次结构转变,CV峰归属需谨慎:氧化还原峰不仅反映过渡金属氧化态变化,还与相变、多相共存及动力学有关。下文结合CV与XANES综合分析。原位Mn K边XANES(图2A、B)表明,低于3 V的氧化峰归属于Mn3⁺/Mn4⁺氧化(图1)。随Mn含量从F5M5降至F8M2,Mn的CV氧化峰相对面积逐次减少约50%,F8M2中几乎消失。但据Mn K边位移,所有样品中Mn均似完全氧化至Mn4⁺。放电时Mn K边相对原始态向低能移动,归因于Na缺失及原始材料中部分氧化Mn态,故放电时(Na≈1)Mn逐渐还原为Mn3⁺。结果与Zhou等一致。四种材料在3.3–3.5 V间均有CV氧化峰,归属Fe3+/Fe4+氧化还原:该电压范围内Fe K边线性偏移,Mn K边不变。随铁含量从F5M5增至F8M2,拟合CV峰相对面积从15.8%增至60.9%(图1C)。F6M4在3.23 V处附加小峰也归属Fe3+/Fe4+,将在相演化部分讨论。所有材料在高电位CV中均有一氧化峰(首次循环尤为显著),表明部分不可逆氧化还原。F5M5中此前归因于氧氧化,原位XANES证实:约4.0 V前Fe K边线性变化,Mn K边平台;超过4.0 V后Fe K边变化速率改变,Mn K边轻微变化,表明氧氧化占主导,边移源于Fe/Mn配位几何变化。随铁含量增加,F5M5中两个高电位峰(3.83 V、4.14 V)合并为一个宽峰,包含Fe3+/Fe4+与氧氧化共同贡献,称为Fe-O峰(图1虚线)。
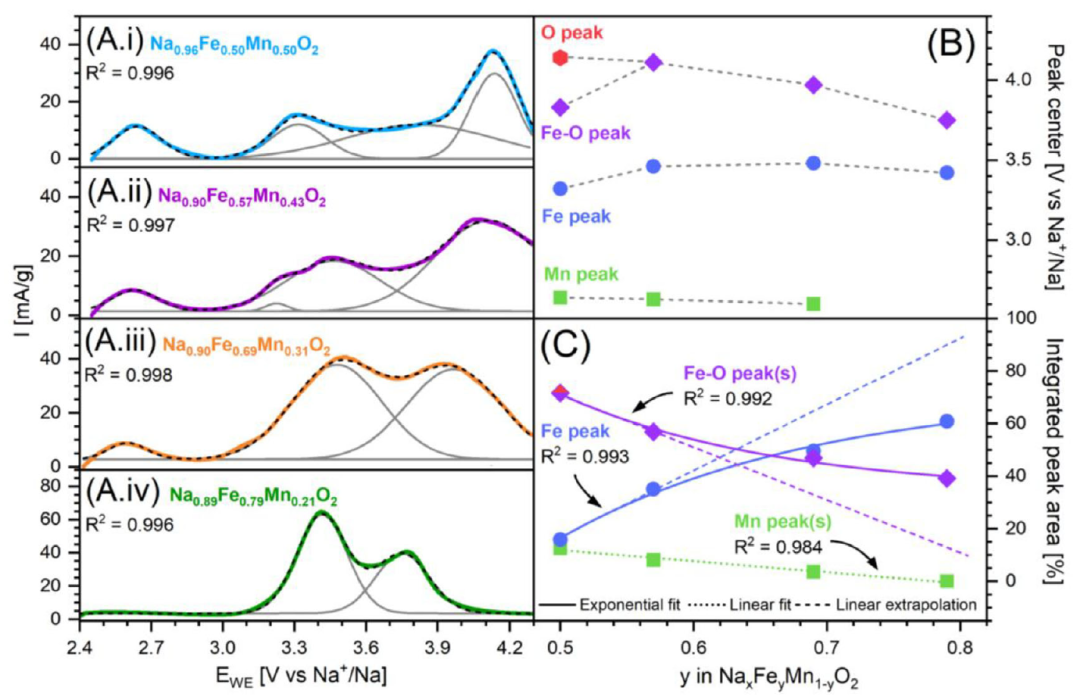
图1. (A) (i) O3-Na0.96Fe0.50Mn0.50O2、(ii) O3-Na0.90Fe0.57Mn0.43O2、(iii) O3-Na0.89Fe0.69Mn0.31O2 和 (iv) O3-Na0.89Fe0.79Mn0.21O2 在首次充电时的循环伏安曲线数据。(B) 随着铁含量增加,CV峰中心位置的变化趋势;(C) CV峰相对面积(占总积分面积的百分比)的变化趋势。
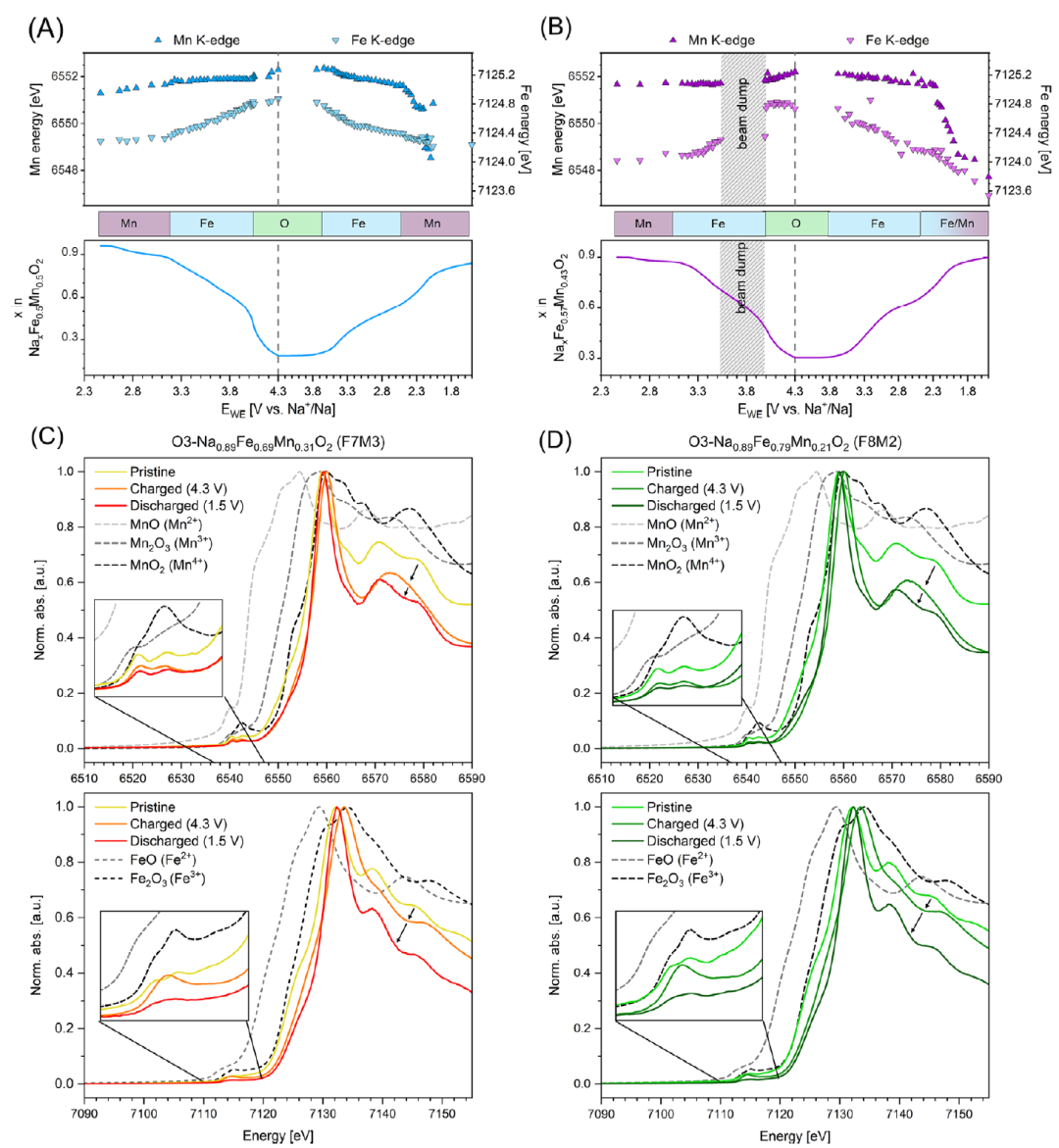
图2. (A和B) 在Na+/Na电位下,Mn和Fe K边原位XANES导数的拟合边缘位置随电位变化的关系,(C) O3-Na0.89Fe0.69Mn0.31O2 和 (D) O3-Na0.89Fe0.79Mn0.21O2的原始、充电(至4.3 V)和放电(至1.5 V)电极的Mn(上)和Fe(下)K边非原位XANES数据。
从F5M5到F8M2,Fe-O峰持续向更低电位偏移(图1B)。更高铁含量增强Fe 3d与O 2p空间重叠,提高Fe-O共价性,提升氧为主的能级能量,降低氧化电位。类似效应见于含铁层状氧化物和富锂材料。随铁含量增加,Fe3+/Fe4+和Fe-O CV峰更尖锐,与单一TM类型主导、周围原子变化小的结构一致。F8M2氧化还原特性类似NaFeO2,首次充电时出现约3.3 V和3.8 V两个电压平台。提高Fe:Mn比例增加铁相关氧化过程相对锰的总贡献,但Fe3+/Fe4+与Fe-O峰相对贡献亦变化:高铁含量时前者逐渐主导(图1C)。另一趋势:Mn相关氧化电荷大致随Mn含量线性增长,而Fe3+/Fe4+氧化电荷不随Fe含量线性变化,实测峰面积系统小于线性预期,且偏差随Fe增加而增大——虽Fe3+/Fe4+贡献增大,但参与该过程的铁比例下降。
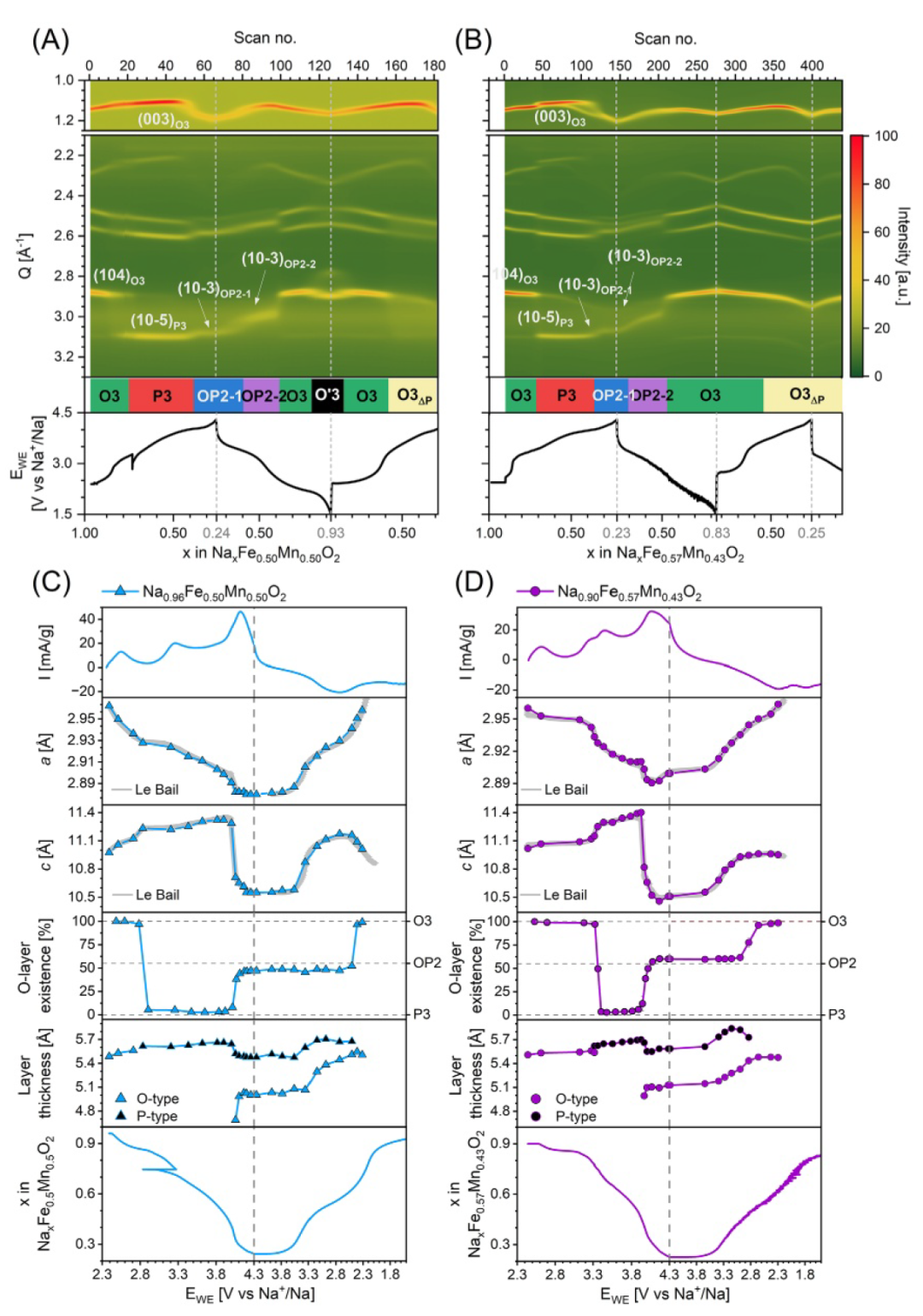
图3. 原位PXRD和恒电流充放电曲线的概览图,显示了电位随插层钠含量x的变化关系,分别对应于(A) O3-Na0.96Fe0.5Mn0.5O2 和 (B) O3-Na0.9Fe0.57Mn0.43O2,并与(C)和(D)中CV及FAULTS精修结果相对应。
XANES数据可估算充电态铁氧化程度。Boivin等指出,Li0.5Mn0.5O2充电至4.5 V时铁平均氧化态仅约+3.3(K边位移0.55 eV);完全氧化为Fe4+则位移达1.45 eV。本研究充电至4.3 V时Fe K边最大位移约0.8 eV,表明铁为Fe3+/Fe4+混合态,非完全氧化。据Fe3+/Fe4+CV峰积分电荷,F5M5、F6M4、F7M3、F8M2中该峰(<3.5 V)分别占理论铁容量的15.8%、33.6%、54.9%、45.6%(为铁氧化量下限)。Fe-O峰(>3.7 V)容量分别占理论铁容量的75.7%、68.3%、54.5%、29.4%。F5M5至F7M3两峰容量和超理论铁容量90%,F8M2为75%。这些数值代表铁氧化上限;但XANES表明所有充电态铁为混合价,故Fe-O峰必含显著氧氧化还原贡献。
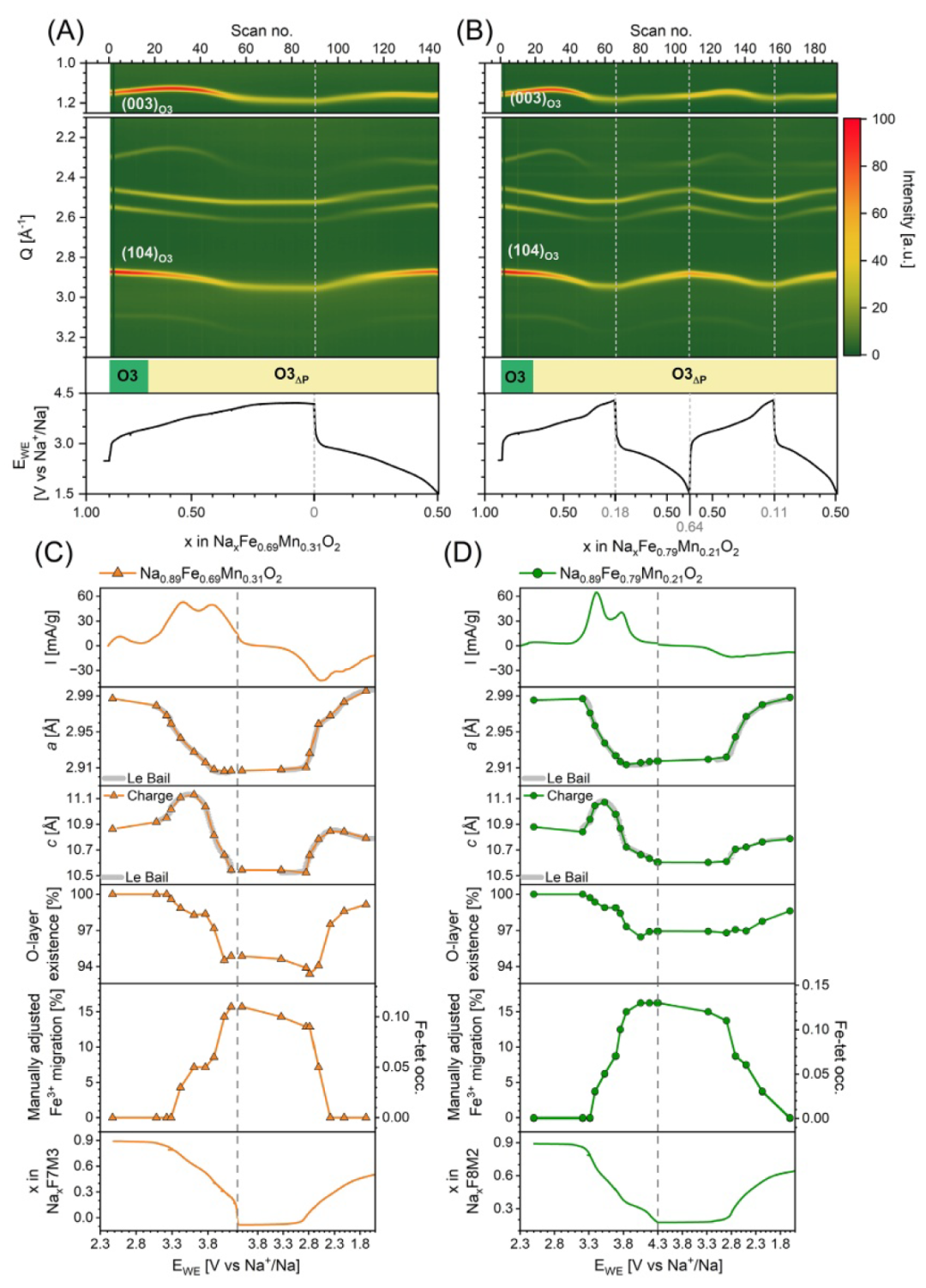
图4原位PXRD和恒电流充放电曲线的概览图,显示了电势随插层钠含量x的变化关系,其中(A)为O3-Na0.89Fe0.69Mn0.31O2,(B)为O3-Na0.89Fe0.79Mn0.21O2,分别对应(C)和(D)中CV及FAULTS精修结果的相关数据。
为了关联氧化还原特性与结构变化,我们利用原位粉末X射线衍射(PXRD)研究Na⁺脱嵌过程中O3-NaxFeyMn1₋yO2的结构演变(图3和图4)。各组成的首次循环衍射图谱经FAULTS精修,O3、P3及完全充电相的精修结果见支持信息。F5M5和F6M4表现出高度相似的结构行为,与先前对F5M5的分析一致。OP2-1和OP2-2相是堆垛层错变体,其中O型和P型层交替排列,形成小畴;X相更无序,O型和P型畴增大,占结构75%以上。O’3相是O3的单斜畸变 ,仅在F5M5中明显,但F6M4显示早期迹象。结构差异与CV相关:F5M5中O3→P3相变发生于~2.8 V(对应Mn3+/Mn4+氧化尾部);F6M4需部分Fe氧化激活相变,发生于~3.3 V(对应CV中3.23 V小峰)。Mn对P相有稳定作用,这与单金属α-NaFeO2中无相变一致。
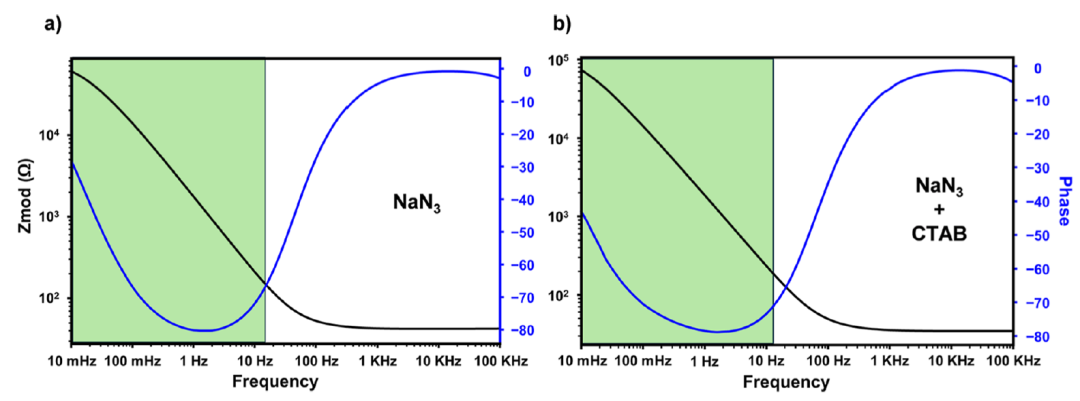
图5. 100 mM叠氮化物(a)和1 mM CTAB、100 mM叠氮化钠混合物(b)的EIS数据,显示出两者之间具有显著的相似性。绿色方框表示进行VibREIS检测的频率范围。
在F6M4中,P3→OP2-1相变发生在约3.9 V(时间戳~0.45),对应Fe3+/Fe4+峰(3.46 V)的高电位尾部及宽泛的Fe-O混合氧化峰(4.11 V)。F5M5中该相变延迟至约4.0 V(~0.41),与Fe-O氧化峰重合。放电时OP2-2→O3相变在F5M5和F6M4中分别发生在约2.5 V(~0.62)和2.8 V(~0.49),凸显锰稳定P型堆叠的关键作用。O3→P3相变伴随a轴收缩、c轴膨胀(源于层间O-O排斥增强)。P3→OP2-1时c轴显著收缩:层厚分析表明仅O型层收缩,P型层无明显变化。由于Fe向四面体位迁移仅可能发生在O型NaO2层中,这强烈表明Fe迁移发生在P3→OP2-1相变期间。原位XANES中Fe前缘微弱可逆增强也支持Fe具有非中心对称配位(如四面体)。PXRD还显示,在F6M4(F5M5程度较小)P3→OP2-1时,Q≈2.5 Å⁻1处出现(003)峰分裂,归因于O型和P型堆垛序列共存。F5M5的a轴在4.0–4.3 V呈平台,F6M4则轻微膨胀,与从过渡金属氧化向氧氧化转变一致,且Fe3+迁移可导致a轴轻微膨胀。F6M4中OP2相的O型层比例(~60%)高于F5M5(~50%),因四面体Fe3+促进O型层形成。放电后,两者分别保持OP2-1相至约3.3 V和3.4 V,OP2-2相至约2.5 V和2.7 V,随后直接转变为O3相,放电态结构与原始材料接近,表明高度可逆。
从F7M3和F8M2的原位PXRD数据(图4)可见,其相演化与F5M5、F6M4显著不同。FAULTS精修表明,引入堆垛层错后O3模型能全程描述衍射数据,未发生向P3的完全转变。富铁材料仅达到少量P型比例(F7M3为7%,F8M2为4%),形成O3Δp相。O3→P3相变在约3.7 V和4.1 V(F7M3)及约3.5 V和4.0 V(F8M2)处停滞,对应Fe3+/Fe4+及Fe-O峰上限。伴随层错形成,Fe3+迁移急剧增加(O型层四面体位点占据率上升),通过PXRD峰强变化追踪。原位XANES(图2C、D)支持:原始态为八面体Fe3+特征,充电态变为单一强峰(四面体/Fe4+/Fe3+),放电后部分可逆。
【工作总结】
本研究揭示了铁含量对O3型钠离子正极(以NaxFeyMn1−yO2为模型体系)在钠脱嵌过程中氧化还原行为、铁迁移及结构演变的影响。结合循环伏安法(CV)、原位XANES和原位粉末X射线衍射(PXRD)分析发现:当y = 0.5–0.6时,首次充电中铁迁移有限,电极可完整经历O3→P3→OP2→O3相变序列;而当y ≥ 0.7时,早期铁迁移“钉扎”O型层,抑制O3→P3相变,使材料始终处于堆垛缺陷的O3Δp结构中。高铁含量导致铁更早、更广泛迁移,并使高电位氧化还原反应向更低电位偏移。铁迁移在早期循环中部分可逆,但随着循环增加可能累积或不可逆捕获。这些发现为富铁O3型电极强烈倾向于O3结构、可逆容量较低提供了机理依据,并强调了对堆垛层错敏感的精修方法在解析高压层状氧化物正极中互生长和层错结构的重要价值。
原标题:《【复材资讯】最新AFM揭示O3型NaxFeyMn1−yO2正极材料中相演变、铁迁移与氧氧化还原的相互作用》