Sotos综合征:从分子机制到临床诊疗的全面解析

Sotos综合征(Sotos syndrome),又称“大脑性巨人症”,是一种以儿童期过度生长、特征性颅面畸形、智力发育障碍为核心表现的罕见常染色体显性遗传性疾病,也是最常见的过度生长伴智力障碍综合征之一。该病于1964年由Sotos首次系统描述,2002年被证实由NSD1基因单倍剂量不足致病,随着分子遗传学技术的发展,其致病机制、临床表型谱、诊断与管理策略均已形成完善体系。本文结合最新研究进展对Sotos综合征进行科普。

01
Sotos综合征致病基因NSD1的
结构与生理功能
(一)基因定位与基本结构
NSD1(Nuclear receptor SET domain-containing protein 1)基因定位于人类染色体5q35.3,全长包含23个外显子,编码含2696个氨基酸的核受体结合SET结构域蛋白,属于组蛋白甲基转移酶家族,别名包括KMT3B、ARA267等。
NSD1蛋白具有多个高度保守的功能结构域,是其发挥生物学功能的核心基础:SET结构域:位于蛋白C端,是组蛋白甲基转移酶的催化核心,负责介导甲基基团向组蛋白尾部转移;SAC结构域:与SET结构域相邻,协同增强甲基转移酶活性,共同调控组蛋白修饰;PHD结构域:共5个植物同源结构域,为锌指样基序,参与染色质调控与蛋白-蛋白相互作用;PWWP结构域:共2个,介导染色质结合与蛋白相互作用,参与基因转录调控;C5HCH结构域:参与染色质重塑,与PHD结构域共同维持染色质稳态;NID⁻/NID⁺ᴸ结构域:位于蛋白N端,为核受体相互作用结构域,兼具转录共激活与共抑制功能。
图1 NSD1基因
Tatton-Brown, Katrina, and Nazneen Rahman. Sotos syndrome. European journal of human genetics : EJHG vol. 15,3 (2007): 264-71. doi:10.1038/sj.ejhg.5201686
(二)生理功能:表观遗传调控的核心分子
NSD1是关键的表观遗传调控因子,核心功能为催化组蛋白甲基化修饰,调控基因转录,参与胚胎发育、细胞增殖分化、生长发育等多个生理过程:
1. 组蛋白甲基化催化:特异性催化组蛋白H3第36位赖氨酸二甲基化(H3K36me2) 和组蛋白H4第20位赖氨酸甲基化(H4K20me),其中H3K36me2是其核心修饰产物,为转录激活相关的表观标记;
2. 增强子调控:通过qPHD-PWWP模块识别增强子区域的H3K18ac修饰,富集于活性增强子,作为转录共激活因子促进RNA聚合酶Ⅱ暂停释放,调控发育相关基因的特异性表达;
3. 染色质稳态维持:拮抗多梳抑制复合体2(PRC2)介导的H3K27me3抑制性修饰,维持染色质开放状态,保障生长发育相关基因的正常转录;
4. 发育调控:在胎儿脑、肾、骨骼肌、肺等组织中高表达,调控神经系统、骨骼系统、泌尿生殖系统的正常发育,是胚胎着床后发育的必需基因。
正常情况下,NSD1通过表观遗传修饰精准调控生长相关基因的表达,维持机体生长发育平衡;一旦其功能丧失,将导致生长调控紊乱、神经发育异常,最终引发Sotos综合征。
02
突变机制与基因型特征
(一)核心致病机制:单倍剂量不足
Sotos综合征遵循常染色体显性遗传模式,95%以上病例为新发突变,仅不足5%为家族性遗传。其核心致病机制为NSD1单倍剂量不足:即一个等位基因发生功能丧失性突变,另一个正常等位基因无法代偿,导致NSD1蛋白表达量或活性下降50%,表观遗传调控紊乱,引发疾病表型。
(二)主要突变类型与分子特征
目前已发现超过500种NSD1致病性变异,主要分为基因内突变和染色体微缺失两大类,不同人群突变谱存在显著差异:
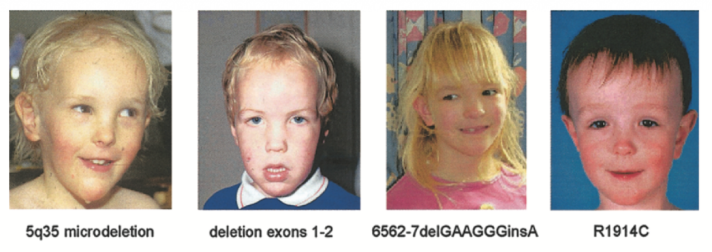
图2 不同NSD1突变患者的典型面部特征
Tatton-Brown, Katrina et al. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. American journal of human genetics vol. 77,2 (2005): 193-204. doi:10.1086/432082
03
临床特征
Sotos综合征的临床表型随年龄动态变化,核心为三联征:特征性颅面畸形、儿童期过度生长、智力/发育障碍,同时累及神经、骨骼、心血管、泌尿等多个系统,疾病谱广泛。
(一)核心临床特征(≥90%患者出现)
1. 特征性颅面畸形
1-6岁最典型,表现为巨头畸形、高宽前额、额颞部发际线后移、眼睑下斜、颧骨潮红、尖下巴;成年后面部变长,下巴更突出,面容仍具特征性。
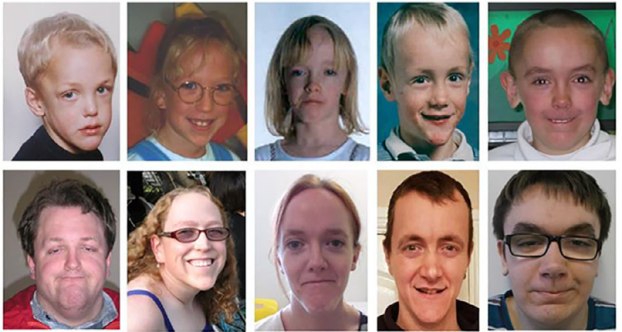
图3 从儿童期到成年期面部外观的变化
Foster, Alison et al. The phenotype of Sotos syndrome in adulthood: A review of 44 individuals. American journal of medical genetics. Part C, Seminars in medical genetics vol. 181,4 (2019): 502-508. doi:10.1002/ajmg.c.31738
2. 儿童期过度生长
出生即表现为身长超标(+2SD以上),儿童期身高、头围持续超97百分位,60%患者超99.6百分位;青春期后生长逐渐正常化,成年女性身高中位数+1.9SD,男性+0.5SD,多回归正常高值范围;巨头畸形可持续至成年。
3. 智力与发育障碍
97%患者存在不同程度学习障碍,其中轻度39%、中度27%、重度16%,仅18%智力正常;运动发育迟缓(独走延迟至15个月后)、语言发育迟缓(2.5岁后说话)为常见首发症状。
(二)主要伴随特征(≥15%患者出现)
1. 骨骼系统:骨龄超前(76%)、脊柱侧弯(30%)、关节松弛/扁平足、膝外翻;
图4 早期脊柱侧弯的影像
Juneja, A, and A Sultan. Sotos syndrome. Journal of the Indian Society of Pedodontics and Preventive Dentistry vol. 29,6 Suppl 2 (2011): S48-51. doi:10.4103/0970-4388.90741
2. 神经系统:新生儿肌张力低下(70%)、癫痫(25%)、头颅MRI异常(脑室扩张、胼胝体发育不良,80%);
3. 新生儿期表现:高胆红素血症、喂养困难、低血糖(70%);
4. 其他系统:心脏畸形(21%,房间隔缺损、动脉导管未闭为主)、肾脏畸形(15%,膀胱输尿管反流、肾盂梗阻)、斜视、便秘。
04
诊断
(一)诊断流程
Sotos综合征诊断采用临床表型+分子检测的金标准流程,2007年欧洲人类遗传学学会发布标准化诊断路径:
1. 临床疑似诊断
满足核心三联征中2项及以上,结合骨龄超前、新生儿肌张力低下等伴随特征,由遗传专科医生评估;
2. 分子确诊检测
一线检测:MLPA(多重连接依赖探针扩增) 筛查5q35微缺失及NSD1部分缺失;
二线检测:Sanger测序/高通量测序 检测NSD1基因内点突变、移码突变等;
3. 确诊标准
临床表型符合+检出NSD1致病性变异(基因内突变或5q35微缺失),即可确诊。
(二)分子检测阳性率
临床确诊Sotos综合征患者中,93%可检出NSD1异常:83%为基因内突变,10%为5q35微缺失,5%为部分基因缺失;仅7%临床典型患者未检出突变,可能为启动子区突变或调控区异常未覆盖。
05
治疗与管理策略
Sotos综合征无根治性治疗,以多学科协作、长期随访、对症支持为核心管理原则,覆盖新生儿期至成年期。
(一)多学科管理团队
包括临床遗传科、儿科、神经内科、骨科、心脏科、肾脏科、康复科、心理科,实现全周期管理。
(二)分阶段临床管理
1. 新生儿期
对症处理:黄疸光疗、喂养支持(鼻饲)、纠正低血糖、治疗胃食管反流;
基线筛查:心脏超声、肾脏超声、头颅MRI,排查先天畸形。
2. 儿童期(0-18岁)
生长监测:每6个月监测身高、头围、骨龄,无需生长抑制治疗,青春期后自然缓解;
神经康复:尽早开展语言治疗、运动康复、认知训练,改善发育迟缓;
并发症管理:癫痫规律抗癫痫治疗;脊柱侧弯定期X线检查,必要时支具/手术矫正;心脏、肾脏畸形专科干预;
行为干预:针对自闭症、焦虑、冲动行为,开展心理疏导、行为矫正,必要时药物辅助。

图5 定期脊柱侧弯检查
3. 成年期
常规随访:每年监测血压、脊柱、听力、牙齿,评估关节、淋巴水肿情况;
生活支持:轻度障碍者支持就业、独立生活;重度障碍者提供长期照护;
遗传咨询:告知生育风险,建议产前诊断。
(三)遗传咨询与产前诊断
1. 再发风险:父母表型正常的先证者,再发风险<1%(无生殖腺嵌合报道);患者生育,子女患病风险50%;
2. 产前诊断:妊娠11-13周绒毛活检、16-22周羊水穿刺,行MLPA或测序检测NSD1突变,实现产前确诊。
(四)肿瘤筛查
无需常规肿瘤筛查:整体肿瘤风险<3%,仅儿童期神经嵴肿瘤、骶尾部畸胎瘤风险略增,无特异性筛查指征;成年肿瘤风险与普通人群一致。
05
结语
Sotos综合征作为NSD1单倍剂量不足导致的表观遗传性疾病,是连接分子遗传学与临床表型的经典范例。从NSD1的表观遗传调控功能,到突变谱与表型关联,再到标准化诊断与多学科管理,该病的研究已形成完整体系。
目前,分子检测是确诊的金标准,多学科长期管理是改善预后的核心,表观遗传靶向治疗是未来突破方向。随着精准医学的发展,早期诊断、个性化干预将进一步提升患者生存质量;同时,对NSD1表观调控机制的深入研究,也将为其他过度生长综合征、神经发育障碍疾病的研究提供重要参考。
对于临床医生而言,需强化对Sotos综合征特征表型的识别,尽早开展分子检测;对于患者家庭,需通过遗传咨询明确疾病本质,坚持长期随访与康复干预。多方协作,才能为Sotos综合征患者提供全生命周期的医疗支持。
参考文献
[1] Ocansey, Sharon, et al. Sotos Syndrome. GeneReviews®, edited by Margaret P Adam et. al., University of Washington, Seattle, 17 December 2004.
[2] Baujat, Geneviève, and Valérie Cormier-Daire. Sotos syndrome. Orphanet journal of rare diseases vol. 2 36. 7 Sep. 2007, doi:10.1186/1750-1172-2-36.
[3] Sun, Zhen et al. Chromatin regulation of transcriptional enhancers and cell fate by the Sotos syndrome gene NSD1. Molecular cell vol. 83,14 (2023): 2398-2416.e12. doi:10.1016/j.molcel.2023.06.007
[4] Tatton-Brown, Katrina et al. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. American journal of human genetics vol. 77,2 (2005): 193-204. doi:10.1086/432082
[5] Harris, Jacqueline R, and Jill A Fahrner. Disrupted epigenetics in the Sotos syndrome neurobehavioral phenotype. Current opinion in psychiatry vol. 32,2 (2019): 55-59. doi:10.1097/YCO.0000000000000481
[6] Foster, Alison et al. The phenotype of Sotos syndrome in adulthood: A review of 44 individuals. American journal of medical genetics. Part C, Seminars in medical genetics vol. 181,4 (2019): 502-508. doi:10.1002/ajmg.c.31738
[7] Tatton-Brown, Katrina, and Nazneen Rahman. Sotos syndrome. European journal of human genetics : EJHG vol. 15,3 (2007): 264-71. doi:10.1038/sj.ejhg.5201686
[8] Priolo, Manuela et al. Further delineation of Malan syndrome. Human mutation vol. 39,9 (2018): 1226-1237. doi:10.1002/humu.23563
[9] Juneja, A, and A Sultan. Sotos syndrome. Journal of the Indian Society of Pedodontics and Preventive Dentistry vol. 29,6 Suppl 2 (2011): S48-51. doi:10.4103/0970-4388.90741
[10] Neeman, Bar et al. Sotos Syndrome: Deep Neuroimaging Phenotyping Reveals a High Prevalence of Malformations of Cortical Development. AJNR. American journal of neuroradiology vol. 45,10 1570-1577. 3 Oct. 2024, doi:10.3174/ajnr.A8364
[11] Zhang, Yong-Ling et al. Sotos syndrome: A study of antenatal presentation. European journal of obstetrics, gynecology, and reproductive biology vol. 279 (2022): 1-4. doi:10.1016/j.ejogrb.2022.10.006
[12] Masunaga, Yohei et al. Sotos syndrome with marked overgrowth in three Japanese patients with heterozygous likely pathogenic NSD1 variants: case reports with review of literature. Endocrine journal vol. 71,1 (2024): 75-81. doi:10.1507/endocrj.EJ23-0502
撰写 宋扬 研究生
原标题:《【科普文章】Sotos综合征:从分子机制到临床诊疗的全面解析》