【复材资讯】ACS Energy Letters:揭示高浓电解质中极限电流的电导驱动原因


【研究背景】
下一代电解质材料必须能够承受高电流密度,以实现快速充放电速率,这对于提高电池性能至关重要。电解质的研究通常侧重于最大限度地提高离子电导率。然而,电解质的性能取决于电流通过时自然形成的盐浓度梯度的大小。这些梯度的大小不仅取决于电导率,还取决于其他传输和热力学参数:迁移率、扩散系数和盐活度系数。浓度梯度的影响最早由亨利·桑德于1901年指出。在电池中,电镀电极处氢气泡的形成被正确地解释为盐耗尽的标志,即电极附近缺乏工作离子。当工作离子浓度在任一电极处接近0时的电流密度即为极限电流。在实际应用中,极限电流对通过电池的施加电流设定了上限,因为当施加电流超过极限电流时,会出现电压曲线发散的情况。极限电流因此是电解质优化的一个强有力的指标。在简化模型中,假设传输参数与浓度无关,浓度分布呈线性;实际中这些参数均与浓度相关,导致分布非线性,但整体定性趋势一致。稀溶液条件下,极限电流随浓度升高而增大;而在浓溶液区,受溶解度限制,极限电流随浓度升高而减小,呈非单调变化。
【工作简介】
在此,斯坦福同步辐射光源实验室Christopher J. Takacs(通讯作者)团队利用原位X 射线透射成像技术,测量了锂-铟对称电池中聚合物电解质在超过限制电流时的时空盐浓度分布。通过浓度分布的测量,能够绘制出包含欧姆分量在内的时空电位分布。正极电导率的急剧下降导致电位差的增大,而非热力学溶解度极限。这一现象是由电导率定的,并且与维持浓度梯度相关的过电位有关。相关成果以“Conductivity-Driven Origin of the Limiting Current in Concentrated Electrolytes”为题发表在《ACS Energy Letters》上。
【研究内容】
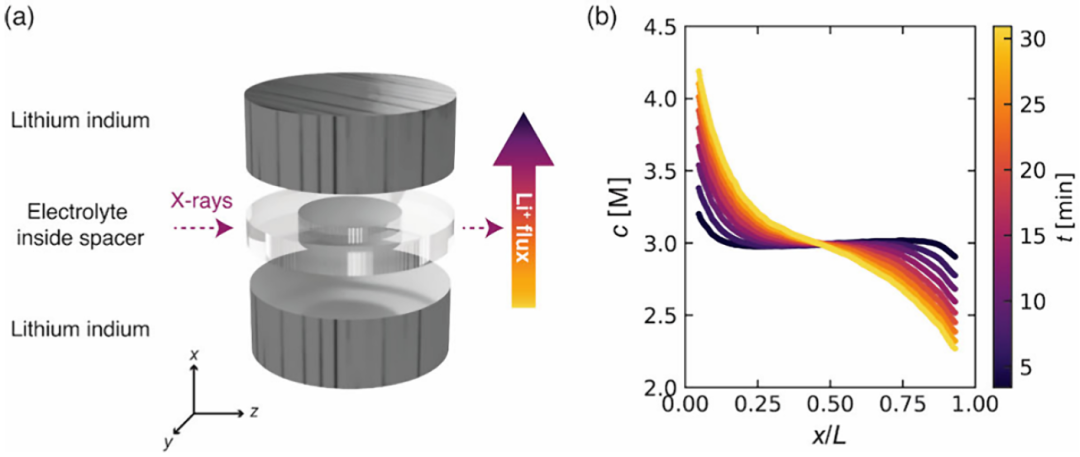
图1.X 射线透射成像装置示意图。
本研究中所考察的聚合物电解质由聚氧化乙烯(PEO)与双(三氟甲烷磺酰)亚胺锂(LiTFSI)盐混合而成。电解质中LiTFSI 的平均浓度以摩尔浓度cavg或摩尔比ravg=[Li+]/[ethylene 表示。作者研究了在cavg=2.98 M (ravg=0.20的 PEO/LiTFSI电解液中的一个电池,施加的电流密度为1.18 mA cm-2。根据参考文献12中实验报告的极限电流,所施加的电流是极限电流的2.1倍。原位电池的示意图如图1a 所示。恒流步骤持续了32.3分钟,此时测量到的电池电压已升至截止电压。此时关闭电流,电池在开路条件下松弛。图1b 展示了在极化步骤期间电解质中盐浓度随时间变化的X 射线成像测量结果。在t = 3.4 分钟的首次测量中,整个电池的浓度分布基本平坦,浓度为c=2.98 M,而浓度变化仅集中在靠近电极的位置,即x/L=0和x/L=1附近。然而,在t=30.9分钟的最后一次测量时,整个电池内都出现了明显的浓度梯度。在x/L=0附近,盐浓度超过c=4.2 M,而在x/L =1附近则降至c =2.3 M以下。此时观察到梯度的不对称性,靠近剥离电极处的斜率明显更陡。对于后续所有0≤x/L≤1范围内的分析,作者使用Python中的Univariate Spline包,通过三次样条插值将测量的c(x,t)数据外推至边缘。
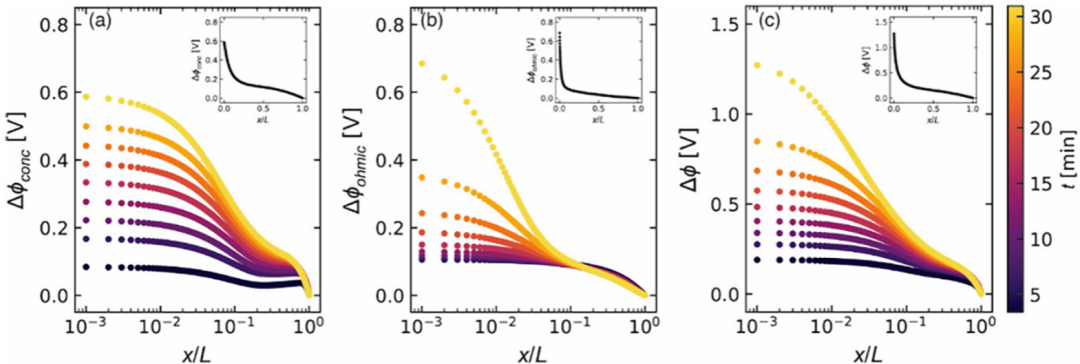
图2.在PEO/LiTFSI中,电流i=1.18 mA cm-2时,局部(a)浓差极化过电位Δϕconc,(b)欧姆压降Δϕohmic以及(c)电解质总电位降Δϕ与电池位置(x/L)和时间(t)的关系。
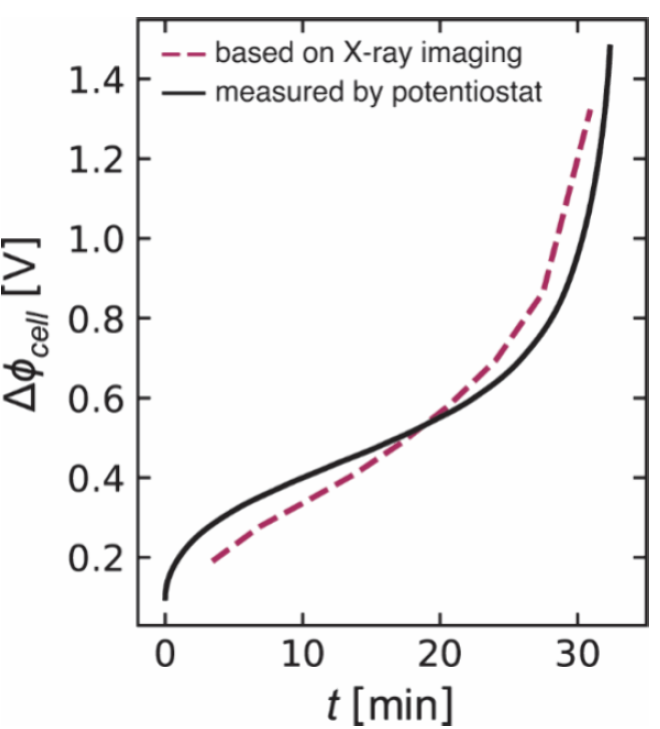
图3.电解质中随时间变化的电位差Δϕ。
在图2中,作者使用x/L 的对数作为横坐标,因为电位的大部分显著变化都局限于x/L 值在0 到 0.1 之间的狭窄范围内。图2中的插图展示了在最终时间点获得的感兴趣的电位(Δϕconc、Δϕohmic和 Δϕ)随x/L 的变化关系,采用的是线性图。这些图与图1c 中所描绘的线性关系大不相同。图2中观察到的非线性反映了热力学和传输参数对浓度的依赖性。
图3中的虚线表示由时空浓度数据得出的电池电势Δϕcell 随时间的变化情况。作者还用实线绘制了通过恒电位仪测量得到的电池电势Δϕexp。将图3中恒电位数据与基于原位c(x,t)数据的预测结果之间的吻合视为对作者分析时空浓度分布方法的有力验证。他们可以将Δϕcell的时间依赖性分解为其可加贡献Δϕcell(欧姆项)和Δϕcell(浓度项),其结果如图4a所示。在所有时间点上,Δϕcell,浓度逐渐增加。在早期阶段(t<4 分钟)主要由Δϕcell(欧姆)主导,因为形成浓度梯度需要一定的时间。在4cell(浓度)主导。然而,在t= 27.5分钟时,出现了一个明显的上升,即Δϕcell(欧姆),并且在t =29分钟时超过了Δϕcell(浓度)。这表明当超过极限电流时,欧姆贡献导致了作者在浓聚合物电解质中观察到的电压偏差。为了进一步说明这一点,作者在图4b 中展示了剥离电极处电解质电导率的时间依赖性。κ随时间的衰减是观察到极限电流行为的原因。在平衡条件下,聚氧化乙烯(PEO)/双(三氟甲烷磺酰)亚胺锂(LiTFSI)混合物在90°C 时表现出复杂的行为,该温度远高于纯PEO 的结晶熔点。通过光学显微镜和差示扫描量热法(DSC)观察到,在浓度高于c =3.62 M时,这些混合物中存在结晶复合物。可能形成EO:Li 比例1、3:1和6:1的结晶复合物。X射线成像数据未显示在极化过中有晶体形成的迹象。也许在作者的极化实验中,成核障碍阻止了晶体的形成。电解质中所能支持的电流受到极限电流的限制,极限电流被定义为电池电压发散时的电流。由于电极处离子耗尽而导致极限电流的原理已得到充分证实。相比之下,高浓度极限下极限电流的起源仍有待确定。此前大多数对此现象的解释都假定电压发散是由于在剥离电极处盐浓度接近溶解度极限所致。
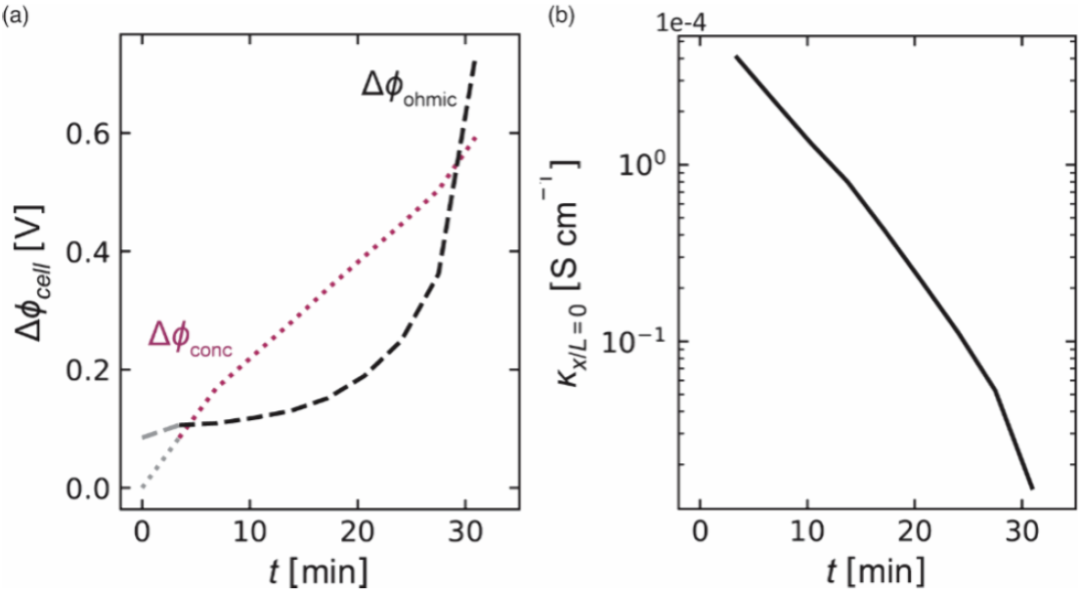
图4.随时间变化的浓度极化过电位Δϕcell。
【工作总结】
作者认为,原位测量浓度梯度对揭示极限电流行为至关重要,而多数高浓度电解质研究未涉及此点。实验采用高浓度PEO/LiTFSI电解质,通过原位X射线成像测量超极限电流下的浓度梯度,测得的电池电位与基于浓度梯度的计算值定量一致。电压发散并非源于溶解度效应引起的浓度过电位发散,而是由剥离电极附近电导率急剧下降导致的欧姆项发散主导。该方法成功解耦了欧姆项与浓度项,确立了电导率在电位增大中的主导作用。高浓度电解质虽在锂金属、高压、锂硫等体系中表现出优异性能,但本工作揭示的摩擦效应为突破下一代电解质材料瓶颈提供了新的设计原则。
原标题:《【复材资讯】ACS Energy Letters:揭示高浓电解质中极限电流的电导驱动原因》