深度学习解决计算量子化学基本问题,探索物质与光如何相互作用

原创 David Pfau等 集智俱乐部

导语
当分子和材料受到大量能量的刺激时,电子可能会跃迁到更高能量的激发态。激发态对理解物质如何与光相互作用至关重要。在此前的研究中,DeepMmind 团队开发了神经网络架构 FermiNet,利用深度学习从第一性原理计算原子和分子的能量。近日,研究团队将这项工作拓展到激发态的计算中,在 Science 上发表用神经网络计算量子激发态的突破性工作,该方法比以往的方法更稳健和通用。
研究领域:量子化学,神经网络,AI for science
David Pfau,James Spencer | 作者
龚铭康 | 译者

论文题目:
Accurate computation of quantum excited states with neural networks
论文地址:
https://www.science.org/doi/abs/10.1126/science.adn0137
2020年发表于 Physical Review Research 上的文章展示了深度学习如何帮助解决现实系统中的量子力学基本方程。这不仅是一个重要的基础科学问题,也可能在未来带来实际应用,使研究人员能够在实验室实际制造之前,通过计算机模拟来原型化新材料和化学合成。
论文题目:Ab initio solution of the many-electron Schrödinger equation with deep neural networks
论文地址:https://journals.aps.org/prresearch/abstract/10.1103/PhysRevResearch.2.033429
神经网络架构 FermiNet(Fermionic Neural Network,费米子神经网络)非常适合模拟大量电子的量子态,电子是化学键的基本组成部分。FermiNet 是第一个利用深度学习从第一性原理计算原子和分子能量的示例,并且其精度足够高,具有实际用途。而我们基于自注意力机制的新架构 Psiformer 迄今为止仍然是最精确的人工智能方法。我们将 FermiNet 加入到蛋白质折叠、玻璃态动力学、晶格量子色动力学等许多项目中,希望在人工智能研究中开发的工具和理念能够帮助解决基础科学问题。
提到“量子力学”,你可能感到困惑。这个词汇让人联想到薛定谔的猫,这只猫悖论般地既可以是活着的,也可以是死去的,还有那些既是粒子又是波的基本粒子。在量子系统中,像电子这样的粒子不像经典描述那样有一个确切的位置。相反,它的位置是由一个概率云描述的——它分布在所有可能存在的地方。这种反直觉的状态让理查德·费曼(Richard Feynman)宣称:“如果你认为你理解了量子力学,那你就不理解量子力学。”
尽管这种诡异的现象让人迷惑不解,但该理论的核心可以简化为几条简单的方程。其中最著名的是薛定谔方程,它在量子尺度上描述粒子的行为,正如牛顿运动定律描述我们熟悉的人类尺度下物体的行为一样。虽然对这个方程的解释可能会引发无尽的困惑,但其数学部分相对易于处理,这也促使教授们在面对学生尖锐的哲学提问时常常会说:“Shut up and calculate!”(“闭嘴,算就完了!”)
这些方程足以描述我们周围所有熟悉的物质在原子和原子核层次上的行为。它们的反直觉性质导致了各种奇异现象:超导体、超流体、激光和半导体的存在都是量子效应的结果。而即便是普通的共价键——化学的基本构成单元——也是电子量子相互作用的产物。
这些规则在 1920 年代被提出后,科学家们意识到他们第一次拥有了一个关于化学如何运作的详细理论。理论上,他们只需为不同的分子建立这些方程,计算出系统的能量,便可以知道哪些分子是稳定的,哪些反应会自发发生。然而,当他们实际着手计算这些方程的解时,发现他们只能精确地计算出最简单的原子(氢原子)的解,而几乎无法处理其他任何原子,它们都过于复杂。
“物理学和整个化学领域的数学理论所必需的基本物理定律已经完全为人所知,唯一的困难是这些定律的精确应用会导致方程过于复杂而无法求解。因此,开发应用量子力学的近似实用方法成为当务之急。”
——保罗·狄拉克(Paul Dirac),量子力学奠基者,1929年
但许多人接受了狄拉克的挑战。物理学家们很快建立了数学技术,能够近似描述分子键和其他化学现象的定性行为。这些方法基于对电子行为的近似描述。在这种描述中,每个电子被分配到一个特定的轨道,轨道给出电子在原子核附近任何一点被发现的概率。每个轨道的形状依赖于所有其他轨道的平均形状。由于这种“平均场”描述将每个电子仅分配到一个轨道上,因此它并没有全面地展现电子的实际行为。尽管如此,它仍足以在大约 0.5% 误差内估算分子的总能量。
不幸的是,0.5% 的误差对于实际工作的化学家来说仍然不够有用。分子键中的能量只占系统总能量的极小一部分,正确预测一个分子的稳定性通常可能只取决于总能量的 0.001%,或者大约 0.2% 的“关联”能量。例如,在丁二烯分子中,电子的总能量接近每摩尔 100,000 千卡,但不同分子形状之间的能量差异仅为每摩尔 1 千卡。这意味着如果你想正确预测丁二烯的自然形态,那么所需的精度就相当于以毫米为单位测量足球场的宽度。
二战后,随着数字计算的出现,科学家们开发了许多超越这种电子“平均场”描述的计算方法。虽然这些方法通常由各种缩写词表示,但它们大致可以归为在精度和效率之间进行权衡的方法的不同极端。一端是精确的计算方法,其计算复杂度随电子数量呈指数级增长,因此除最小的分子外,这些方法都无法实际应用。另一端则是线性扩展的方法,但精度不高。这些计算方法对化学实践产生了巨大影响——1998年诺贝尔化学奖颁发给了这些算法的创始人。
费米子神经网络
尽管现有的计算量子力学工具种类繁多,我们认为仍然需要一种新方法来解决高效表示的问题。即使是最粗略的方法,最大的量子化学计算也仅能处理数万电子,而经典的化学计算技术如分子动力学可以处理数百万个原子。
经典系统的状态可以很容易地描述——我们只需跟踪每个粒子的位置和动量。描述量子系统的状态则困难得多,需要为每种可能的电子位置构型赋予一个概率。这种构型编码在波函数中,波函数为每种电子构型分配一个正或负数,波函数的平方给出了系统处于该构型中的概率。
所有可能构型的空间是巨大的——如果你尝试用每维100个点来表示,那么对于硅原子来说,可能的电子构型数量将超过宇宙中的原子数量。这正是我们认为深度神经网络(deep neural networks)可以发挥作用的地方。在过去几年里,使用神经网络表示复杂的高维概率分布取得了巨大进展。我们现在知道如何高效且可扩展地训练这些网络,并猜测鉴于这些网络能够在人工智能问题中拟合高维函数,或许它们也可以用来表示量子波函数。
此前的研究展示了现代深度学习如何用于解决理想化的量子问题。我们希望使用深度神经网络来解决化学和凝聚态物理中更现实的问题,这意味着我们必须在计算中考虑电子。
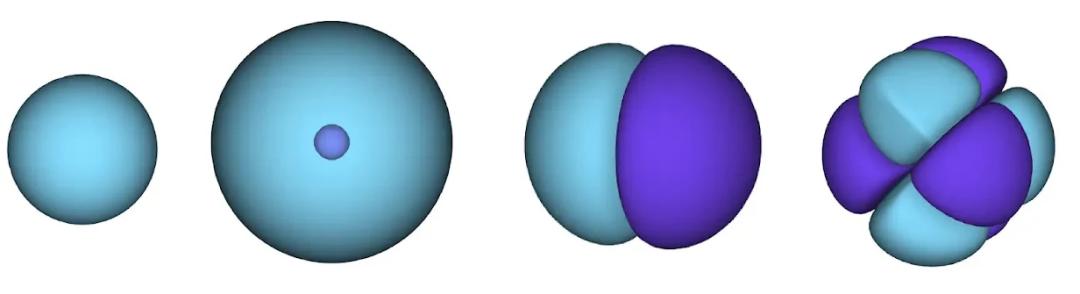
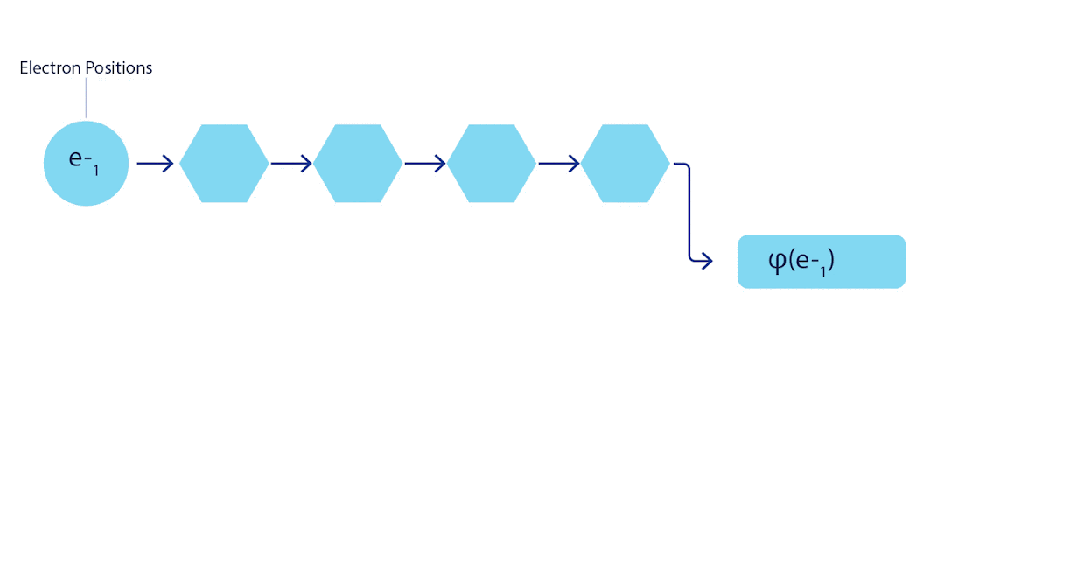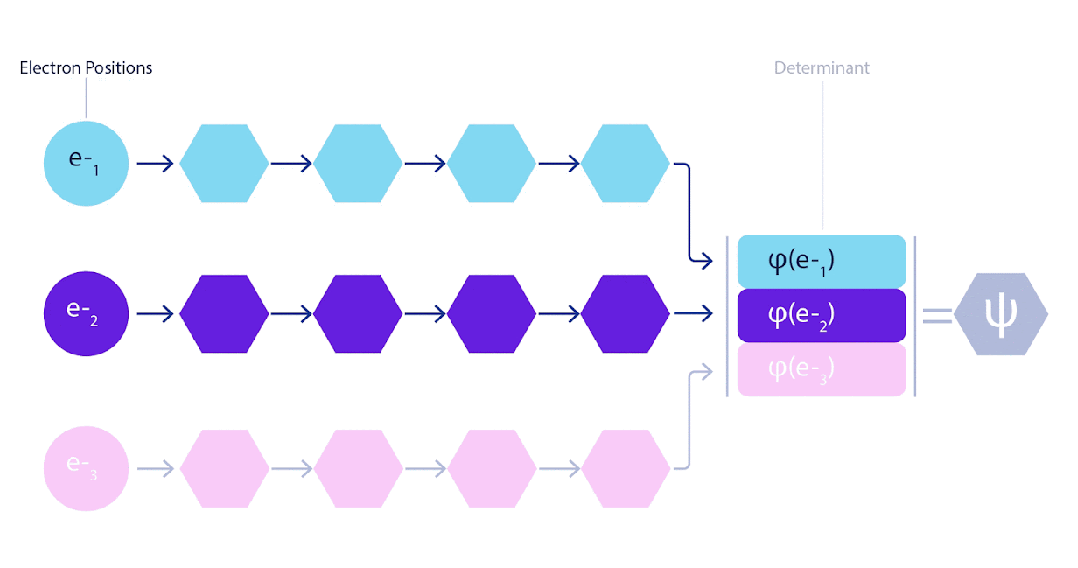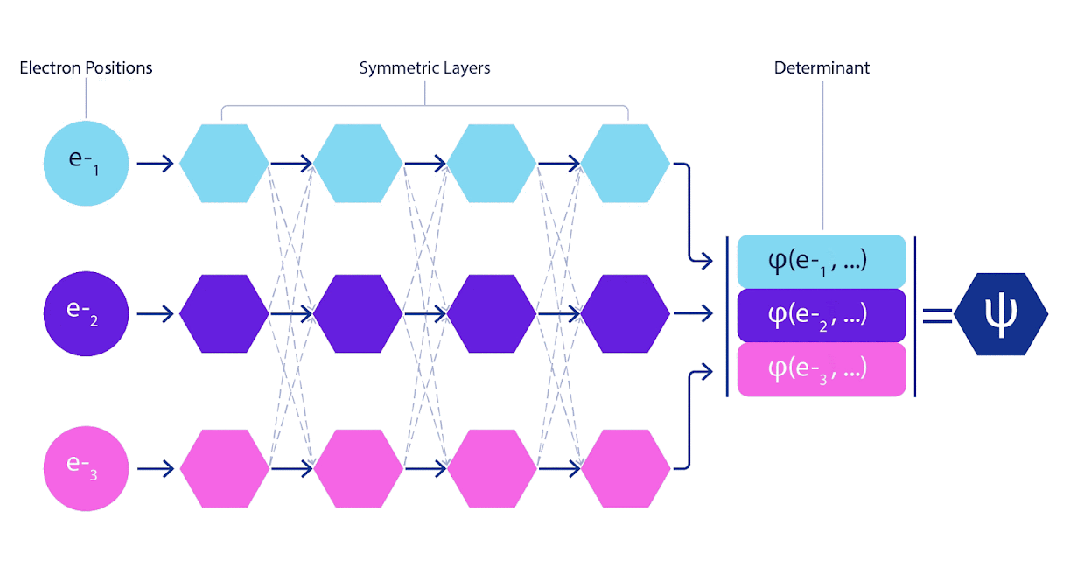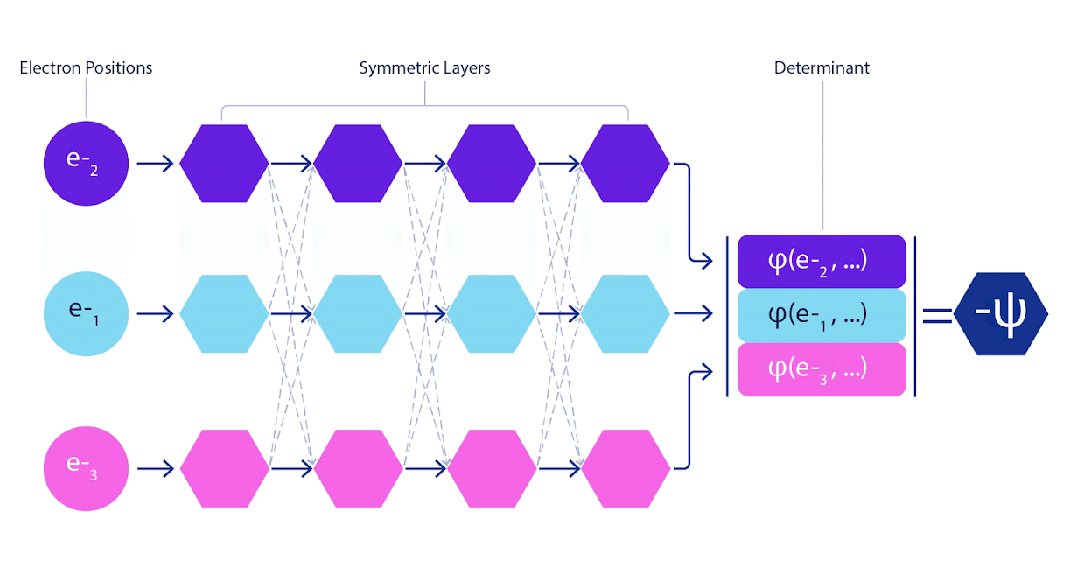
处理电子时只有一个小难点。电子必须遵守泡利不相容原理(Pauli exclusion principle),不能同时处于同一个空间。这是因为电子是一种称为费米子(fermions)的粒子,这类粒子包括大多数物质的组成部分:质子、中子、夸克、中微子等。它们的波函数必须是反对称的。如果交换两个电子的位置,波函数就会乘以-1。这意味着如果两个电子重合,波函数(以及该构型的概率)将为零。
这意味着我们必须开发一种对输入具有反对称性的神经网络,我们称之为 FermiNet。在大多数量子化学方法中,反对称性是通过一种称为行列式(determinant)的函数引入的。矩阵的行列式具有这样的性质:如果交换两行,输出就会乘以-1,正如费米子的波函数一样。因此,你可以取一组单电子函数,对系统中的每个电子进行评估,并将所有结果打包成一个矩阵。该矩阵的行列式便是一个适当的反对称波函数。这种方法的主要局限性在于,所得出的函数——称为斯莱特行列式(Slater determinant)——并不具备广泛适用性。
真实系统的波函数通常要复杂得多。改善这种情况的典型方法是采用大量斯莱特行列式的线性组合——有时可能是数百万个或更多——并基于电子对进行一些简单的修正。即便如此,这也可能不足以精确计算能量。
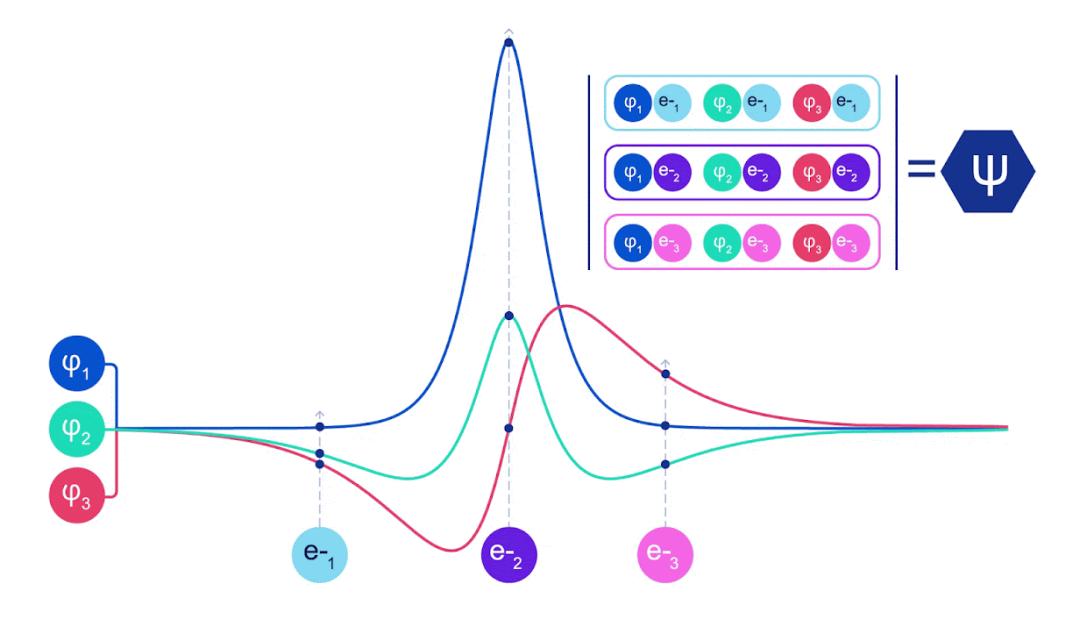
深度神经网络通常在表示复杂函数方面比线性组合的基函数更加高效。在 FermiNet 中,这是通过将每个进入行列式的函数设定为所有电子的函数来实现的。这远远超越了仅使用一电子和二电子函数的方法。FermiNet 为每个电子提供了独立的信息流。如果这些信息流之间没有相互作用,该网络的表现力将不会超过传统的斯莱特行列式。
为了超越这一点,我们在网络的每一层对所有信息流的信息进行平均,并将这些信息传递给下一层的每个信息流。这样,这些信息流就具有了正确的对称性质,从而创建一个反对称函数。这类似于图神经网络在每一层汇聚信息的方式。与斯莱特行列式不同,FermiNet 是通用函数逼近器,至少在神经网络层足够宽时是这样的。这意味着,如果我们能够正确地训练这些网络,它们应该能够拟合接近精确的薛定谔方程解。
我们通过最小化系统的能量来拟合 FermiNet。要精确地做到这一点,需要在所有可能的电子构型下评估波函数,因此只能进行近似计算。我们随机选择一些电子构型,局部评估每种电子排列下的能量,汇总每种排列的贡献并最小化它,而不是最小化真实能量。这被称为蒙特卡洛方法(Monte Carlo method),因为它有点像赌徒不断掷骰子。虽然它是近似的,但如果我们需要更精确,可以再次掷骰子。
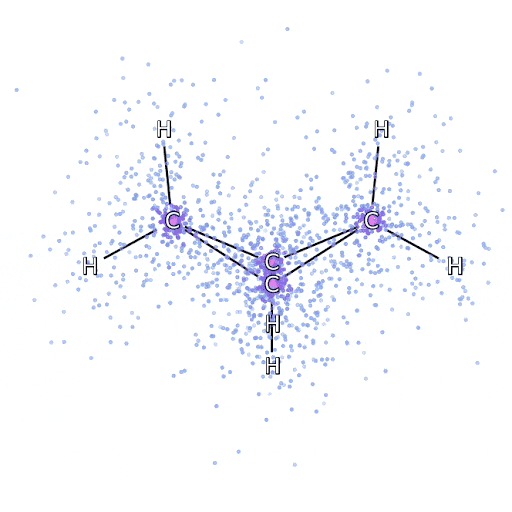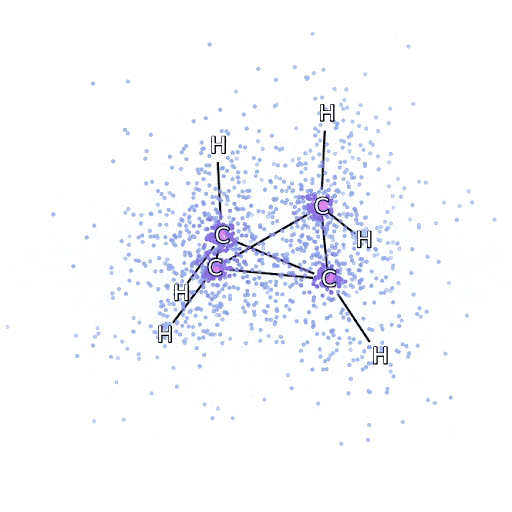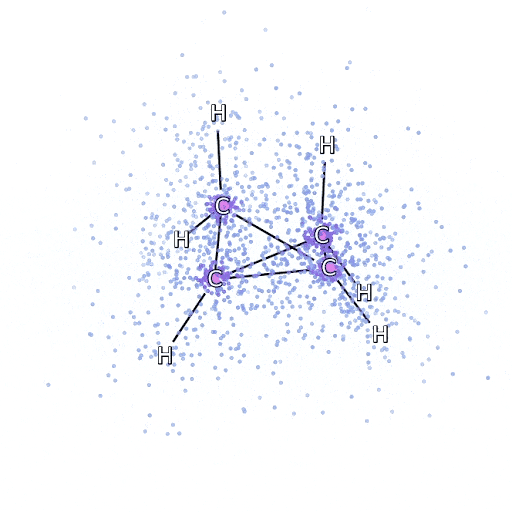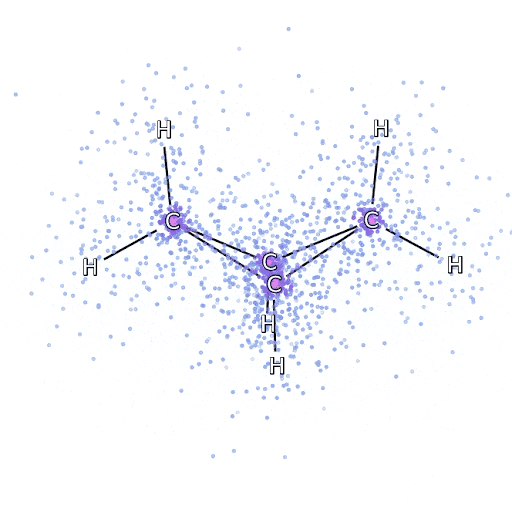
由于波函数的平方给出了观察到粒子在任意位置排列的概率,因此最方便的做法是直接从波函数中生成样本——本质上是在模拟观察粒子的行为。虽然大多数神经网络是从某些外部数据中训练的,但这里用来训练神经网络的输入数据是由神经网络自身生成的。这意味着除了电子绕原子核“跳舞”的位置外,不需要其他训练数据。
这一基本思想被称为变分量子蒙特卡洛法(variational quantum Monte Carlo,简称VMC),自60年代以来一直存在,通常被认为是一种计算系统能量的廉价但不太精确的方法。通过用 FermiNet 取代基于斯莱特行列式的简单波函数,我们在所有研究的系统上显著提高了这一方法的精度。
为了确保 FermiNet 代表了当前技术的进步,我们首先研究了一些简单且被深入研究过的系统,比如元素周期表第一周期的原子(从H到Ne)。这些都是小型系统——电子数量少于10——足够简单,可以用最精确(但规模呈指数增长)的方法来处理。
FermiNet 的表现远远超过了相对的 VMC 计算——通常能将相对于指数扩展计算的误差减少一半甚至更多。在较大的系统中,指数扩展方法变得难以处理,因此我们改用耦合簇方法(coupled cluster method)作为基准。该方法在分子处于稳定构型时效果很好,但在键被拉伸或断裂时表现不佳,而这对理解化学反应至关重要。虽然其扩展方式比指数扩展好得多,但我们使用的特定耦合簇方法的规模依然随着电子数量的七次方增长,因此它只能用于中等尺寸的分子。
我们将 FermiNet 应用于逐渐增大的分子,首先从氢化锂开始,逐步研究到双环丁烷,这是我们研究的最大系统,包含30个电子。在最小的分子中,FermiNet 捕获了耦合簇能量与单一斯莱特行列式能量之间惊人的 99.8% 差异。在双环丁烷中,FermiNet 仍捕获了 97% 或更多的关联能量,对于这样一种简单的方法来说,这是一个巨大的成就。
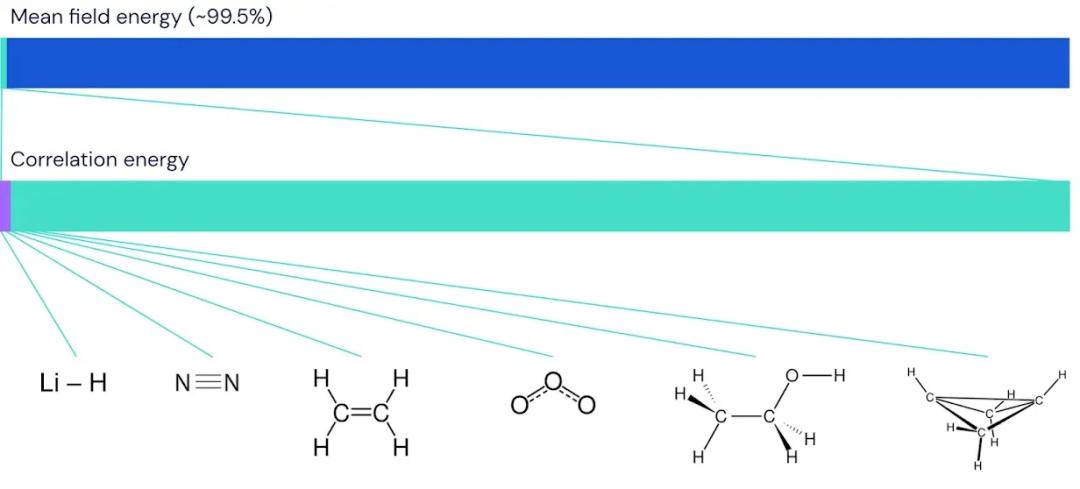
虽然耦合簇方法在稳定分子中表现良好,但计算化学的真正前沿是在理解分子的拉伸、扭曲和断裂过程上。在这些方面,耦合簇方法常常会遇到困难,因此我们必须与尽可能多的基准进行比较,以确保得到一致的答案。
我们研究了两个拉伸系统的基准:氮分子(N2)和含有10个原子的氢链(H10)。氮分子特别具有挑战性,因为每个氮原子贡献三个电子。而氢链则对理解电子在材料中的行为很有意义,比如预测某种材料是否会导电。
在这两个系统中,耦合簇方法在平衡状态下表现良好,但在键被拉伸时出现了问题。传统的 VMC 计算整体表现不佳,但无论键长如何变化,FermiNet 都是调查的最佳方法之一。
计算激发态的新方法
2024年8月,我们在《科学》杂志上发表了这项工作的下一阶段成果。我们的研究提出了一种解决计算量子化学中最困难挑战之一的方案:理解分子在受到刺激时如何从基态转变到激发态,或反过来。
FermiNet 最初专注于分子的基态,即给定原子核周围的电子最低能量构型。但当分子和材料受到大量能量的刺激时,例如暴露于光照或高温下,电子可能会跃迁到更高能量的构型——即激发态。
激发态对理解物质如何与光相互作用至关重要。吸收和释放的能量精确值为不同的分子和材料创造了独特的“指纹”,这会影响从太阳能电池板和 LED 到半导体、光催化剂等各种技术的性能。它们还在涉及光的生物过程(如光合作用和视觉)中发挥关键作用。
精确计算激发态的能量比计算基态能量要困难得多。即使是基态化学中的黄金标准方法(如耦合簇),在激发态的计算中也会出现几十倍的误差。尽管我们希望将 FermiNet 的工作扩展到激发态,但现有方法不足以让神经网络与最先进的方法竞争。
我们开发了一种计算激发态的新方法,比以往的方法更稳健和通用。该方法可以应用于任何类型的数学模型,包括 FermiNet 和其他神经网络。它的工作原理是通过在一个扩展的系统中引入额外的粒子来找到基态,从而可以对现有的优化算法进行较少的修改后使用。
我们在广泛的基准测试中验证了这项工作,并获得了非常有前景的结果。在一种小而复杂的分子——碳二聚体(carbon dimer)中,达到了平均绝对误差(MAE)为 4 meV 的结果,比之前的黄金标准方法 20 meV 更接近实验结果。我们还在计算化学中一些最具挑战性的系统中测试了该方法,这些系统中有两个电子同时被激发,结果发现误差在 0.1 eV 范围内,这与迄今为止最复杂的计算相匹配。
我们将最新的工作开源,并希望研究界能基于我们的方法,探索物质与光相互作用的新奇方式。
原文链接:https://deepmind.google/discover/blog/ferminet-quantum-physics-and-chemistry-from-first-principles/
原标题:《深度学习解决计算量子化学基本问题,探索物质与光如何相互作用》