线粒体自噬(mitophagy)是重要的线粒体质量及数量调控机制,与发育、癌症、衰老和神经退行性病变等生理病理状况高度相关。现已知两类哺乳动物mitophagy通路:由PINK1-Parkin介导的泛素依赖途径,以及由BNIP3, NIX和FUNDC1等介导的受体依赖途径。然而,人们对mitophagy调控机制及其生理病理意义仍有待探索。突出的表现是,缺失已知mitophagy通路的小鼠(如Pink1-/-、Parkin-/-、Bnip3-/-等)表型微弱,与普遍认知的mitophagy重要性有明显差距。
2023年12月26日,北京生命科学研究所/清华大学生物医学交叉研究院蒋辉实验室在Molecular Cell在线发表了题为A mitophagy sensor PPTC7 controls BNIP3 and NIX degradation to regulate mitochondrial mass的论文。该论文与实验室于2023年7月3日发表在EMBO Journal的A mitochondrial SCF-FBXL4 ubiquitin E3 ligase complex degrades BNIP3 and NIX to restrain mitophagy and prevent mitochondrial disease论文一起,发现了一条感知并抑制mitophagy的PPTC7-SCFFBXL4通路,阐明了它对线粒体数量和代谢稳态的调控作用,并揭示过度mitophagy导致的线粒体数量减少是线粒体DNA耗竭综合征13(MTDPS13,由FBXL4突变引起的致死性退行性疾病1,2)的致病机理。在该通路中,PPTC7感知mitophagy受体BNIP3和NIX的蛋白水平,进而起始BNIP3/NIX-PPTC7-SCFFBXL4完整复合物的组装,促进SCFFBXL4泛素化和降解BNIP3和NIX,从而抑制mitophagy。该通路对维持细胞线粒体数量、代谢平衡、和个体存活至关重要。


蒋辉实验室长期从事线粒体蛋白稳态调控机制研究(详见BioArt报道:从线粒体外膜蛋白的降解说起),之前报道了一个线粒体外膜复合物UBXD8-VCP介导BNIP3降解,从而抑制mitophagy(详见BioArt报道:EMBO Reports | 蒋辉实验室揭示UBXD8调控凋亡和线粒体自噬的分子机制)。为进一步发掘mitophagy调控机制,研究生曹钰通过靶向线粒体的遗传筛选发现多个mitophagy抑制因子,其中包括FBXL4和PPTC7。鉴于FBXL4突变导致MTDPS13的病因未明,作者首先研究FBXL4。
作者发现FBXL4是线粒体外膜跨膜蛋白,且与Skp1和Cullin1组成SCFFBXL4 E3泛素连接酶。遗传筛选发现敲除mitophagy受体BNIP3和NIX可以抑制FBXL4-KO细胞里异常激活的mitophagy。细胞和生化实验表明,SCFFBXL4通过泛素化降解BNIP3和NIX来抑制mitophagy。而病人来源的FBXL4突变抑制SCFFBXL4组装,导致BNIP3和NIX蛋白积累和mitophagy过度激活。FBXL4突变患者表现出mtDNA耗竭、线粒体数量减少、酸中毒、神经和肌肉退行性病变等严重症状。与FBXL4突变病人类似,Fbxl4-/-小鼠表现出BNIP3和NIX蛋白积累、mitophagy失控、线粒体数量减少、代谢紊乱和围产期死亡等严重表型。更重要的是,在Fbxl4-/-小鼠中敲除Bnip3或Nix可以挽救线粒体数量、恢复代谢平衡并挽救小鼠寿命。
因此,该研究发现了一个线粒体外膜E3泛素连接酶SCFFBXL4及其底物BNIP3和NIX,并揭示过度mitophagy导致的线粒体数量减少是MTDPS13的致病机理。值得一提的是,MTDPS13病症和Fbxl4-/-小鼠表型是目前已知由mitophagy紊乱引起的最严重表型。
PPTC7调控FBXL4介导的BNIP3和NIX降解
鉴于BNIP3和NIX强大且危险的线粒体清除能力,细胞必须牢牢控制其水平,但细胞监控并调节BNIP3和NIX降解的分子机制仍不清楚。研究生孙雨秋研究另一个筛选发现的mitophagy抑制因子PPTC7。前人研究表明PPTC7是定位于线粒体基质的磷酸酶。但令人诧异的是,与敲除FBXL4类似,敲除PPTC7同样导致BNIP3和NIX的积累和过度mitophagy;Pptc7-/-小鼠表现出mitophagy失控、线粒体数量减少和围产期死亡的表型,且该表型可以被Nix敲除所挽救。此外,PPTC7过表达可促进内源BNIP3和NIX被FBXL4降解,而FBXL4过表达则没有效果,提示PPTC7作为上游限速因子调控FBXL4介导的BNIP3和NIX降解。但是,线粒体基质蛋白PPTC7如何调控线粒体外膜上的蛋白降解?
PPTC7前体通过与BNIP3/NIX直接互作,滞留在线粒体外膜
通常情况下,线粒体基质蛋白的前体形式(precursor)在N端定位序列 (targeting sequence) 的引导下进入线粒体,并在线粒体基质切除定位序列,完成成熟过程。作者发现在野生型细胞里,PPTC7以成熟形式(mature form,切除了定位序列)存在于线粒体基质,而在FBXL4-KO细胞里,PPTC7同时具有定位于线粒体基质的成熟形式和定位于线粒体外膜的前体形式。进一步分析表明,FBXL4-KO细胞中积累的BNIP3和NIX导致PPTC7前体出现在线粒体外膜;在野生型细胞中直接过表达BNIP3或NIX也可诱导PPTC7前体出现在线粒体外膜定位。
通过体外重组蛋白实验,作者发现BNIP3和NIX与PPTC7直接互作,并使用交联质谱分析了NIX-PPTC7的互作区域。在截去NIX与PPTC7的直接互作区域后,NIX无法诱导PPTC7前体的外膜定位,表明BNIP3和NIX通过与PPTC7的直接互作,将PPTC7前体定位于线粒体外膜。
弱化的PPTC7定位序列帮助PPTC7在线粒体外膜滞留
有意思的是,作者发现与经典线粒体基质定位序列相比,PPTC7的定位序列长度短且引导能力差,推测该特征削弱了PPTC7前体进入线粒体基质的能力,有利于PPTC7前体被外膜BNIP3/NIX滞留。因此,研究者将PPTC7的定位序列替换成经典序列,发现它使PPTC7全部进入线粒体基质,无法被滞留在外膜,失去调节BNIP3和NIX降解的能力。
外膜PPTC7起始组装BNIP3/NIX-PPTC7-SCFFBXL4复合物
那么外膜PPTC7如何促进SCFFBXL4介导的BNIP3和NIX降解呢?作者发现除与BNIP3/NIX直接互作之外,PPTC7还与SCFFBXL4复合物中的Cullin1直接互作。作者表达纯化了NIX,PPTC7,FBXL4,Cullin1-Rbx1和Skp1的重组蛋白,发现只有在所有蛋白都存在的情况下,这些蛋白才能组装成完整的NIX-PPTC7-SCFFBXL4复合物,缺少NIX,PPTC7,FBXL4任何一个组分都导致复合物散架。
以上结果提供了一个mitophagy的负反馈调控模型:积累在线粒体外膜的mitophagy受体BNIP3和NIX与PPTC7前体直接互作,将其滞留在外膜,起始BNIP3/NIX-PPTC7-SCFFBXL4复合物的组装,从而泛素化和降解BNIP3和NIX,防止mitophagy过度激活(图3)。
禁食情况下,肝脏诱导PPTC7来抑制mitophagy和维持代谢稳态
一个重要的问题是PPTC7是单纯的mitophagy负反馈调控因子,还是也会响应生理信号?鉴于PPTC7过表达能促进BNIP3和NIX降解,作者重点研究了生理信号对PPTC7表达的调节,发现禁食不改变骨骼肌PPTC7的表达,但在肝脏强烈诱导PPTC7。与之相对应,禁食强烈诱导骨骼肌mitophagy,但不改变肝脏mitophagy水平,作者推测肝脏mitophagy受到PPTC7上调的压制。
因此,作者在小鼠肝脏敲除PPTC7,发现禁食导致缺失PPTC7的肝脏丢失大量线粒体。禁食情况下,肝脏负有重要使命-通过糖异生(gluconeogenesis)维持血糖水平。而糖异生需要线粒体提供两个重要支持:ATP和丙酮酸羧化酶(pyruvate carboxylase)。作者发现,敲除肝脏PPTC7导致禁食状态下肝脏ATP缺乏,糖异生受阻,造成严重的低血糖。因此,禁食情况下,肝脏特异上调PPTC7以压制mitophagy,从而维持线粒体数量和代谢稳态(图3)。
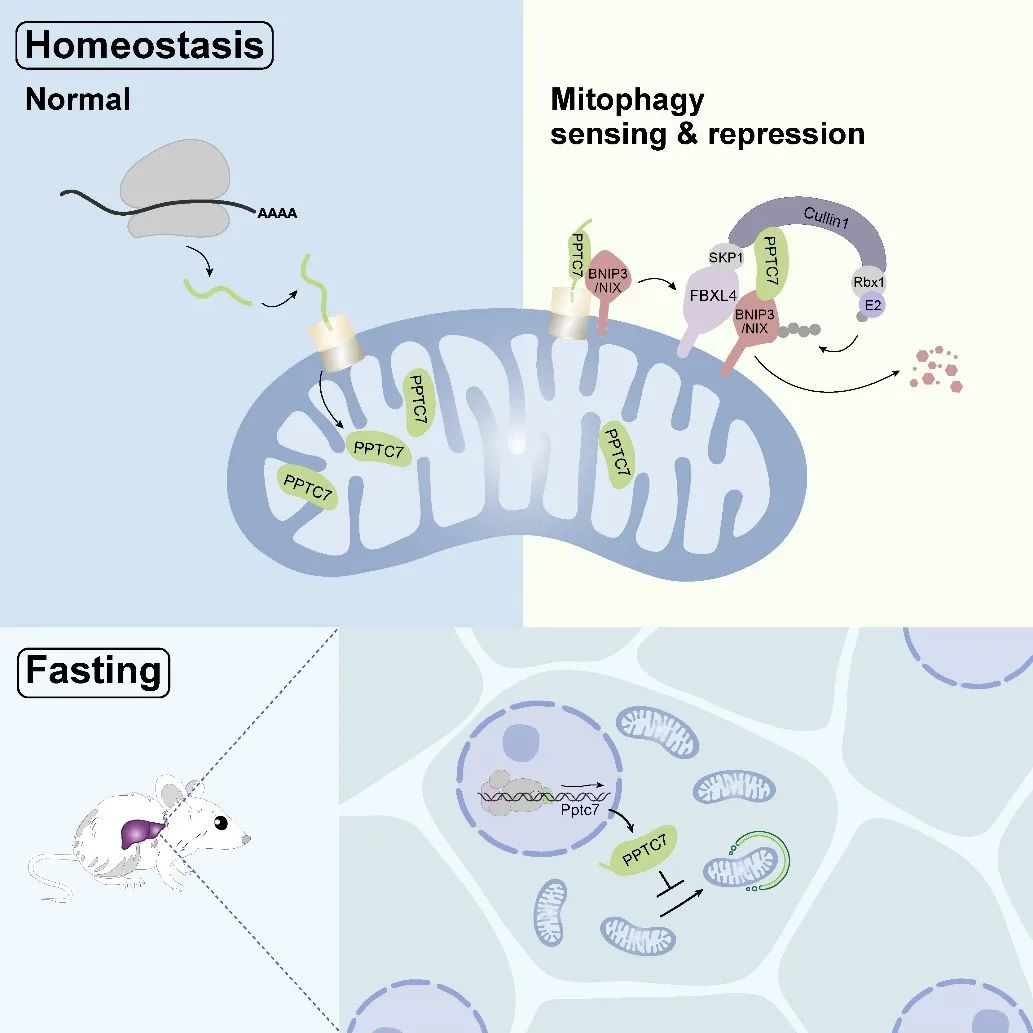
综上,该系列工作报道了一个感知和抑制mitophagy的PPTC7-SCFFBXL4通路。PPTC7作为关键调控因子,能够整合稳态维持和生理信号来调控mitophagy水平,以此维持线粒体数量和细胞代谢稳态。该通路的发现极大增进了我们对mitophagy与线粒体数量控制(mitochondrial mass control)的理解。禁食情况下,肝脏和骨骼肌对mitophagy的差异化调节及其功能研究也丰富了我们对生理情况下组织特异性mitophagy调控的认知。
清华大学生物医学交叉研究院博士生孙雨秋为Molecular Cell论文第一作者,北生所和北师大联合培养的博士生曹钰为EMBO Journal论文第一作者,蒋辉博士为两篇论文的通讯作者。两篇论文其他作者包括蒋辉实验室的郑静、万华云、傅松、刘珊珊和何白羽,北生所董梦秋博士、陈涉博士、马燕博士、李祺博士、王凤超博士、阿达莱提买买提明、曹杨、蔡改红、李琳、吴重阳和王濛。
原文链接:
https://doi.org/10.1016/j.molcel.2023.11.038
https://www.embopress.org/doi/epdf/10.15252/embj.2022113033
制版人:十一
参考文献
1 Bonnen, Penelope E. et al. Mutations in FBXL4 Cause Mitochondrial Encephalopathy and a Disorder of Mitochondrial DNA Maintenance. The American Journal of Human Genetics 93, 471-481, doi:https://doi.org/10.1016/j.ajhg.2013.07.017 (2013).
2 Gai, X. et al. Mutations in FBXL4, Encoding a Mitochondrial Protein, Cause Early-Onset Mitochondrial Encephalomyopathy. The American Journal of Human Genetics 93, 482-495, doi:https://doi.org/10.1016/j.ajhg.2013.07.016 (2013).
原标题:《【学术前沿】Mol Cell & EMBO J丨蒋辉团队揭示感知并抑制线粒体自噬的通路及其对线粒体数量的调控作用》