以下文章来源于老顽童说 ,作者老顽童说

老顽童说
公众号致力于传播衰老相关的前沿科研进展和趣味科普,帮助大家更深入地了解衰老背后的科学故事~

关注我们,获取更多相关资讯
翻译 By 孙晓燕、单贺珍、刘迪、蔺舫民

引言
“岁岁年年花相似,年年岁岁人不同”,在人体内细胞会不断地分裂,不断地替换掉原来的细胞。但这种“轮回”也并非永恒,细胞也遵循着生命的守恒定律,分裂一定次数后,分裂过程就会停滞。那究竟是什么控制了细胞的更新换代呢?2009年的诺奖宣告:“端粒”这一结构在默默地把控细胞宿命。“蜡炬成灰泪始干”,作为染色体保护罩的端粒消耗殆尽之时,细胞也就走向了衰老。由此,“端粒与衰老的关系”成为了科研界的热门,相关研究成果也纷至沓来。2021年1月14日,来自德克萨斯州大学安德森癌症中心的Ronald A. DePinho教授团队在Cell杂志上发表综述文章“Telomeres: history, health, and hallmarks of aging”,全面地阐述了端粒的“来龙去脉”,重点剖析了端粒功能障碍与衰老标志物之间的联系,以及端粒和端粒酶失调在促进衰老、早衰综合征和常见的年龄相关疾病发生中的作用。
通讯作者 Ronald A. DePinho教授(图源网络)
摘要
世界人口老龄化导致的社会和经济负担日趋加重,衰老机制研究刻不容缓。衰老的标志物由多种分子机制与细胞体系组成,它们之间相互关联,共同促进衰老。作者从端粒生物学出发,介绍了端粒功能障碍如何影响多种衰老标志物的分子生物学过程,进一步加剧衰老相关疾病的发展,如神经退行性疾病和癌症。阐明端粒与衰老标志物间的密切联系,将为发现延缓衰老进程与减少衰老相关疾病的干预手段提供参考和借鉴。
上个世纪以来公共卫生和医学的进步使得全球预期寿命急剧增长。联合国报道称,根据目前的趋势,2050年将有21亿人超过60岁,而这将伴随着衰老相关疾病发生率的大幅度增高(如阿尔茨海默氏病,心血管疾病和癌症等)。这些疾病的发生率在60岁及以上人群中每隔5年就会翻一番。如果没有找到有效的医疗干预手段,全球将遭受难以承受的慢性病负担,并造成巨大的社会和经济损失。衰老与慢性病之间的联系促使人们对衰老机制和延缓衰老策略进行基础性研究。
衰老是一个不断发展变化的退行性过程,常伴有组织干细胞耗竭、组织炎症、基质改变、细胞衰老和代谢功能障碍等。这些细胞和组织的变化反映了线粒体、蛋白稳态、细胞间通讯、营养感应、表观遗传学和DNA修复等潜在的异常变化,这些异常变化导致了基因组不稳定和损坏,包括端粒功能障碍。随着对各种衰老相关分子机制更深入的了解,人们意识到端粒功能障碍是驱动衰老及其相关疾病的分子通路的诱因或放大剂。
端粒由重复的核苷酸序列组成,形成“帽结构”,发挥维持染色体完整性的作用。人端粒维持相关的基因发生缺陷时,会引起生殖细胞和体细胞的退行性疾病,如先天性角化病、特发性肺纤维化、溃疡性结肠炎等。端粒等位基因功能丢失或可诱导的基因编辑小鼠加快了端粒功能障碍和衰老、早衰综合征、慢性炎症以及退行性疾病等相关性的研究。内源性端粒酶的重新激活可逆转具有端粒功能障碍小鼠的过早衰老。鳉鱼和斑马鱼都是端粒生物学研究的模型,它们的端粒长度与人类相似,而其端粒功能障碍的表型同啮齿类动物模型更相似些。
作者综述了端粒研究的进展和现状,重点介绍了端粒功能障碍与衰老标志物的联系,阐述了端粒和端粒酶在衰老、早衰综合征、年龄相关退行性疾病(如神经退变和癌症)中似乎无所不在的作用,以及端粒与衰老机制之间的密切联系如何助力于延缓衰老和预防衰老相关疾病的干预手段的发展。
端粒和端粒酶研究历史
端粒的概念诞生于二十世纪三十年代,当时McClintock和Muller推断玉米和黑腹果蝇的染色体末端存在着独特的结构,并认为这对于防止染色体末端融合至关重要。Muller结合了希腊语telos(意为“结束(end)”)和meros(意为“部分(part)”)创造了端粒(telomere)一词,意为“结束部分(end part)”(图1)。1961年,人们证实了人类胎儿细胞具有有限的复制潜能,只可以复制50到60次,被称为“海夫里克极限”或复制衰老。二十世纪七十年代初,Olovnikov(1973)和Watson(1972)由线性DNA复制的不对称现象引入了“末端复制问题”,推测由于末端RNA引物的去除,每次细胞分裂中落后的染色体末端会发生DNA丢失,导致染色体逐渐缩短。
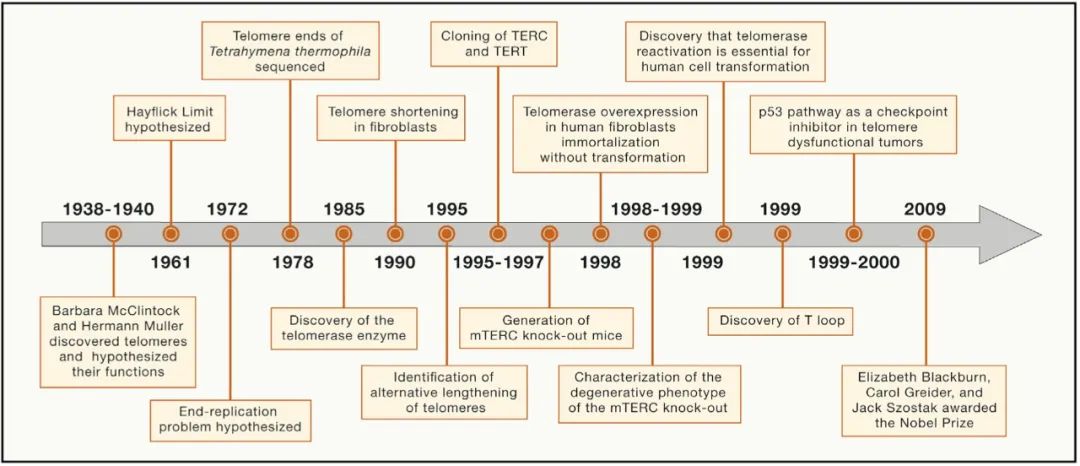
图1 端粒研究时间线
1978年,Blackburn和Gall对纤毛原生动物噬热四膜虫的rDNA进行测序,发现了六核苷酸串联重复序列5’-CCCCAA-3’(互补链为:3’-TTGGGG-5’)组成的末端。1985年,Greider和Blackburn发现了一种能够将DNA重复序列添加到染色体末端,延长端粒长度的新型酶,现在被称为端粒酶。1989年,Greider和Blackburn又从嗜热假单胞菌中克隆得到了端粒酶的RNA。1990年,Harley等人发现并证实了端粒磨损与人原代细胞培养过程中的复制衰老平行发生,表明端粒缩短会触发海夫里克极限。通过激活DNA损伤检查点得到过表达TRF2突变形式(TRF2∆B∆M)的人成纤维细胞。利用此细胞,科研人员在2003年发现了端粒减损可以诱导永久性细胞周期停滞。
下一个重大突破发生在1996年,端粒酶被证明是一种作用于3’突出端的核糖核蛋白。随后,人端粒酶逆转录酶(human telomerase reverse transcriptase, hTERT)和人端粒酶RNA组分(human telomerase RNA component, hTERC)也被成功克隆出来。第一只TERC敲除小鼠在1997年诞生。在随后的几年中,单独敲除TERC和TERT或结合早衰以及癌症相关等位基因突变的小鼠的实验证实了端粒功能障碍可导致早衰、癌症和各种退行性疾病。这些小鼠模型证明了完整的端粒可维持基因组稳定性、组织干细胞储备、器官系统稳态和正常寿命。在1998年,一项里程碑式的研究表明,hTERT表达增强可以赋予人原代细胞无限复制的潜力,这些细胞包括成纤维细胞、视网膜色素上皮细胞和血管内皮细胞,这些细胞在培养过程中始终保持正常的核型且没有出现恶性增殖的现象。
回顾端粒研究史,尽管当时已经发现端粒酶在癌症中上调,但仍不确定端粒酶的功能。当增加TERT表达会出现与经典癌基因促进原代人细胞恶性转化一样的效果时,其在细胞转化中的作用才得到进一步证实。同时,癌症的遗传模型表明p53依赖的DNA损伤应答的状态决定了端粒缩短在体内发挥促进还是抑制癌症的作用。进一步分析mTERC-/-; p53+/-小鼠发现,小鼠上皮癌表现出人源化肿瘤谱特性,即发生了人类癌症基因组典型的染色体重排和不可逆转位现象。因此,DNA损伤信号失活(即p53缺乏)的背景下发生端粒损耗的小鼠实验揭示了老年人中易发生上皮癌的一种机制,以及此类癌症的细胞遗传学特征发生根本性改变的原因。
TERC和TERT敲除小鼠模型验证了端粒在衰老中的作用,并确定了驱动衰老过程的核心信号通路。首先,这些模型确定端粒功能障碍会导致加速衰老的迹象和表型,即寿命缩短,外貌老化,组织干细胞储备减少,器官萎缩以及应对压力、损伤和再生需求的能力下降。这些模型也突显了端粒功能障碍在早衰综合征和帕金森氏病中的重要性。其次,对TERC-/-小鼠后代不同组织进行的转录组学分析揭示了衰老相关的p53-PGC途径,整合了三种先前独立的衰老理论:遗传毒性应激(端粒功能障碍)、氧化损伤和线粒体衰退。最后,可诱导的TERT小鼠模型证明,内源性端粒酶的激活可逆转小鼠的早衰表型。此外,用腺病毒递送端粒酶可改善急性心肌梗死后的心脏功能,增强肌肉协调性以及肾、肝功能,降低胰岛素抵抗和皮下脂肪消耗,增加骨矿物质密度,延长寿命,但不会引起癌症发生率的增加。几十年来,这些多元化的模型系统共同确定了端粒的分子生物学特征及其在健康和疾病中的作用。
遮蔽体(Shelterin)-端粒酶复合物
端粒可以维持染色体的完整性,这对于维持物种的生命周期和繁殖至关重要。端粒末端保护的功能从较低等的多细胞生物(如嗜热链球菌)到较高等的生物(包括人类)都是进化上保守的。在结构上,端粒由TTAGGG的串联重复序列组成,该串联重复序列含有几千到几万个碱基,并在3’末端形成富含鸟嘌呤核苷酸的75到300个核苷酸的单链(图2B)。1999年,Griffith等人首次发现了3’突出端向后折叠,形成一个套索状结构,并将其称为T环(图2A)。
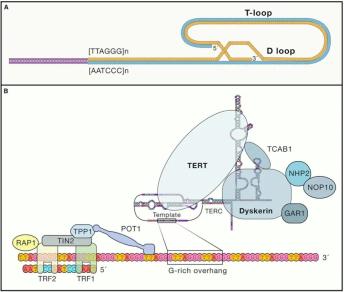
图2 脊椎动物端粒/端粒酶复合物
(A)端粒末端D环和T环结构。
(B)端粒酶结构。NOP10, 核仁蛋白家族A; TIN2, TERF1相互作用核因子; TPP1, 端粒保护蛋白1; TRF1, 端粒重复结合因子1; TRF2, 端粒重复结合因子2; POT1, 端粒保护1; RAP1, TERF2互作蛋白; TCAB1, 端粒酶Cajal体蛋白1; GAR1, 核仁蛋白家族A。
端粒隐藏在被称为遮蔽复合物的专门蛋白质中,该复合物是由六个蛋白质亚基组成的多聚体:TRF1、TRF2、TPP1、POT1、TIN2和RAP1。这种端粒的高阶结构可抑制端粒末端的DNA损伤,通过重组或经典/替代性非同源末端连接可以阻止来自融合末端的DNA修复程序,调节端粒酶在末端的进入和活性。相应地,上述复合物中的突变会破坏端粒-遮蔽体复合物,导致末端融合和过早衰老。TRF1的过表达或POT1的下调会损害端粒酶与端粒末端的结合,从而导致端粒缩短。相反,Bloom(BLM)解旋酶在复制过程中执行稳健的双链断裂修复,TRF1的丢失会由于无法募集BLM解旋酶而在端粒DNA中形成常见易碎位点。
在正常组织中,端粒酶在生殖细胞中高度表达,同时也在皮肤、肠道、造血系统、毛发和睾丸的未分化干细胞和祖细胞中表达。分化的细胞,如角质形成细胞、成纤维细胞、骨骼肌细胞、神经元、心肌细胞和精子中TERT表达水平微弱,甚至检测不到。
TERC与小卡哈尔体RNA(small Cajal body RNAs, scaRNA)和小核仁RNA(small nucleolar RNAs, snoRNA)有关。尽管此类RNA由其他基因的内含子编码,但TERC是自身具有启动子的原型基因。TERC RNA以含有5’甲基鸟苷帽和poly(A)尾的前体转录形式存在。这些前体被疾病相关的poly(A)特异性核糖核酸酶(poly(A)-specific ribonuclease, PARN)腺苷酸化,从而促进TERC的成熟和积累。TERC含一个由3个核苷酸组成的H/ACA域,称为卡哈尔体定位域(Cajal body localization, CAB),该序列对于结合端粒酶卡哈尔体蛋白1(telomerase Cajal body protein 1, TCAB1)是必不可少的。TCAB1是将端粒酶转运至钙质体进而转运至端粒末端以及催化其活性所必需的。端粒酶通过其假结/模板结构域(pseudoknot/template domain, PK/T)和CR4/5结构域与TERC结合。多种蛋白质对于全酶的正常运作都是必不可少的,包括dyskerin、NHP2、NOP10和GAR1,它们和其他蛋白质一起构成核心成分。
总之,端粒结构以及端粒酶活性和募集的精确调控,确保了正常细胞中端粒的维持。同时,它们又都容易受到突变和失调的影响,从而导致家族性和散发性疾病。但是,关于端粒酶复合物是如何感应信号并被招募到最短端粒以及不同成分的特定组装顺序等问题仍然没有解决。并且这些过程在正常和肿瘤细胞中调控的差异性机制仍不清楚。最重要的问题是,还需要更深入地了解调控端粒酶表达和功能的调控过程,以确定端粒酶对人类正常衰老以及遗传性和体细胞性退行性疾病发病机理的作用。
端粒失调与细胞衰老标志之间的相关性
端粒维持的有限性被认为是细胞永生化的一大障碍,端粒功能的丧失与年龄相关的适应性降低和癌症诱发的基因组不稳定密切相关。实际上,端粒功能障碍已被描述为衰老的九大标志之一。在本节中,作者详细介绍了端粒在衰老过程中的角色,重点讨论了端粒与衰老其他标志物之间的联系(图3)。此外,还总结了用于研究端粒与衰老、早衰机制之间相互联系的遗传模型系统。
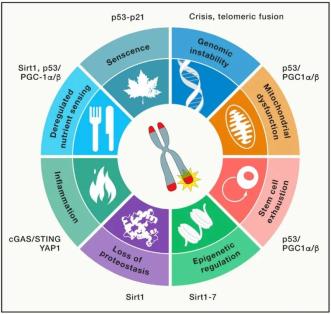
图3 端粒失调与细胞衰老标志之间的相关性
端粒失调驱动各种细胞衰老标志。
细胞衰老
衰老细胞在衰老的组织中积累,并通过各种机制促使衰老和年龄相关疾病的发生。研究最为深入的两个方面包括:组织干细胞(干细胞耗竭,衰老的另一标志)、免疫细胞(免疫衰老)和基质细胞的复制潜能受阻;衰老细胞释放促炎因子(包括但不限于白介素6(interleukin-6, IL-6)和肿瘤坏死因子α(tumor necrosis factor alpha, TNF-α)破坏器官功能。海夫里克极限可以通过激活p53-p19ARF和p16Ink4a-Rb信号通路导致衰老和增殖停滞。尽管已有研究表明,活性氧(reactive oxygen species, ROS)诱导的端粒损伤会导致端粒功能损伤位点(telomere dysfunction-induced focus, TIF)产生,进而诱导衰老。但是,正常衰老过程中端粒功能障碍是否能够激活以及如何激活低增殖能力的基质组织的衰老程序还需要进一步探究。
干细胞耗竭
组织干细胞耗竭是衰老的主要特征。在TERT或TERC功能缺失的小鼠中,持续性地端粒损耗会激活p53依赖的凋亡途径,导致组织干细胞耗竭,诱发器官萎缩,尤其是在自我更新率高、快速增殖的组织中,如皮肤、肠、睾丸、受伤的肝脏和血液等。除了端粒维持,TERT还可能通过TERT的非经典功能(WNT(Wingless-related integration site)途径激活,该途径是调节干细胞稳态的主要通路)影响干细胞生物学功能。具体来说,在小鼠细胞中TERT可以与Brahma相关基因1(Brahma-related gene-1, BRG1)发生相互作用,并作为β-catenin复合物中的辅助因子,导致WNT网络相关基因的上调。
基因组不稳定性
基因组不稳定性是衰老的另一个标志,可导致干细胞耗竭,进而引发炎症。TERC功能缺失细胞和组织的细胞遗传学分析证明端粒功能障碍可加剧染色体的不稳定性,同时也在包括大肠癌在内的多种肿瘤中出现了基因扩增、缺失、易位和后期桥形成的现象。受损或未加帽的端粒会发生末端-末端融合,形成双中心染色体,导致断裂融合桥(breakage-fusion-bridge, BFB)循环、非整倍体和四倍体、易位和扩增,在有丝分裂过程中通过局部超突变(localized hypermutations,)和染色体碎裂(chromothripsis,即clustered chromosomal rearrangements, 簇状染色体重排)造成基因组不稳定。此外,p53的丢失会使细胞免于DNA双链断裂造成的死亡,产生异常的染色体失衡和驱动癌症的不可逆转位。这些染色体异常已在非恶性的老年干细胞中发现,其中突变累积程度与人类组织(包括结肠隐窝和造血系统)年龄密切相关。
线粒体功能障碍
端粒与线粒体紧密相关。线粒体功能随衰老下降,导致能量(ATP)产生减少、细胞内ROS增加。能量产生减少会导致细胞脆性增加,而ROS升高会引发细胞损伤,包括DNA中8-氧鸟嘌呤碱基损伤(端粒富含鸟嘌呤)。线粒体DNA聚合酶亚基γ(polymerase subunit gamma, POLG)突变小鼠相关实验为线粒体功能障碍对衰老的影响提供了的直接证据。这些突变小鼠表现出线粒体数量减少、线粒体形态异常(如线粒体嵴碎片化和外膜破裂),以及过早衰老(如脱发、驼背、体重减轻、皮下脂肪减少、骨密度降低、骨质疏松症、贫血和心肌病)。值得注意的是,这些线粒体功能障碍相关的早衰表型与端粒酶缺乏小鼠,p53过度活化小鼠和PGC1α/β 缺失小鼠的表型相似。考虑到线粒体在衰老中的中心地位,TERC、过氧化物酶体增殖物激活受体g共激活因子1(peroxisome proliferator activated receptor gamma co-activator, PGC1)α/β和POLG功能缺失小鼠的重叠表型使我们能够将端粒,线粒体和氧化防御机制联系起来。已有研究证明TERC缺失的小鼠线粒体功能受损,氧化防御能力降低。此外,对TERC-/-小鼠不同组织的转录组分析显示p53和PGC1α/β的靶基因作用明显,更加使我们确定了三种竞争性衰老理论的共同途径:遗传毒性应激积累,线粒体功能下降,氧化损伤增加。具体而言,端粒功能异常会激活p53,进而抑制PGC1α和PGC1β的表达。PGC1α/β表达降低反过来抑制线粒体生物发生和功能,减少氧化防御相关基因的表达。端粒-p53-PGC1α/β-线粒体的信号回路导致ROS水平升高,进而加剧端粒鸟苷碱基上8-羟基脱氧鸟苷的修饰(图4)。该信号环路构成了一个连接端粒功能障碍,线粒体和氧化应激途径的反馈环,最终导致了衰老加速。
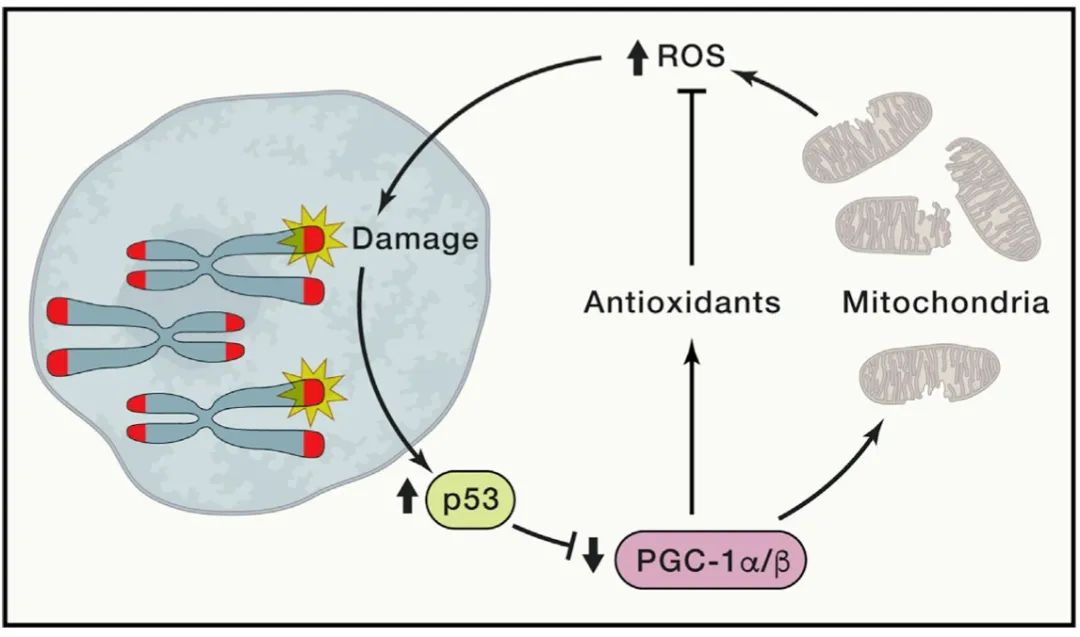
图4 端粒功能障碍导致线粒体缺陷
端粒功能异常损害线粒体功能和氧化防御能力,增加ROS水平,构成端粒-p53-PGC1α/β -线粒体的反馈环。
表观遗传紊乱
年龄相关的表观遗传变化包括局部甲基化增加和整体甲基化减少,H4K16乙酰化、H3K4三甲基化和H4K20三甲基化增加以及H3K9单甲基化、H3K27三甲基化降低。端粒和表观遗传的直接相互作用反映在sirtuins(SIRT)的调控中。SIRT是可以调节寿命和健康寿命的烟酰胺腺嘌呤二核苷酸(NAD +)依赖性脱乙酰基酶(SIRT1–SIRT7)的蛋白质家族。在哺乳动物中,SIRT1可以去乙酰化多个衰老相关的转录因子,包括p53、叉头盒转录因子(forkhead-box transcription factor, FOXO)、PGC1α和核因子kB(nuclear factor kB, NF-kB)。它们是调节应激、代谢和氧化防御相关的关键分子途径。端粒功能障碍诱导p53介导的七个SIRT蛋白表达抑制证明了端粒功能障碍与SIRT(SIRT1和SIRT6)之间的相互作用。此外,端粒功能障碍介导的p53激活会抑制PGC1α/β的表达,进而调控线粒体SIRT3、SIRT4和SIRT5的表达。p53激活也会导致microRNA(miR-34a-5p、miR-26a-5p和miR-145-5p)的转录上调,从而抑制非线粒体SIRT1、SIRT2、SIRT6和SIRT7的翻译。因此,端粒调节的表观遗传网络与衰老密切相关。
蛋白稳态失衡
蛋白质稳态失衡也是衰老标志之一,端粒功能障碍和由此产生的p53介导的SIRT1表达抑制与此相关。蛋白质稳态失衡源于帮助蛋白正确折叠的伴侣蛋白活性下降。热休克蛋白70的相互作用蛋白(heat shock protein 70-interacting protein)CHIP(热休克家族伴侣分子)的羧基末端突变的小鼠表现出加速衰老的表型。在哺乳动物细胞中,SIRT1介导的热休克因子1(heat shock factor-1, HSF-1)的脱乙酰增强了热休克基因(如HSP70)的转录激活。因此,端粒功能障碍引起的SIRT1抑制和由此导致的HSP70水平降低可能是应激时蛋白质稳态失衡,热休克反应下降的原因。蛋白质折叠对于维持神经元稳态十分重要。由于无法通过细胞分裂减少错误折叠蛋白的负荷,有丝分裂后长寿神经元极易受到错误折叠蛋白的影响。蛋白质折叠失调在几种衰老相关神经系统疾病中经常发生,如阿尔茨海默氏病和帕金森氏病,分别表现出错误折叠的b-淀粉样蛋白肽和a-突触核蛋白。错误折叠蛋白的积累逐渐导致神经元死亡和认知障碍。最近研究表明,在阿尔茨海默氏病和帕金森氏病的果蝇、小鼠和大鼠模型中,HSP70的过表达均具有神经保护作用。
营养感应改变
衰老过程中失调的营养感应是进化高度保守的,营养感应途径由IGF-1、mTOR-S6通路、FOXO、AMP激活的蛋白激酶(AMP-activated protein kinase, AMPK)等家族的成员组成。这些途径相互调节,维持代谢稳态。AMPK是营养的中央传感器,可以调节mTOR信号,激活FOXO转录因子和SIRT1。反过来,SIRT1可以激活FOXO和PGC1α,这对于线粒体生物发生至关重要。因此,端粒功能障碍影响代谢的能力源于其对p53的激活以及对PGC1α和SIRT1的抑制。确实,由于糖原异生发生缺陷,端粒功能失调的小鼠不能在禁食条件下维持血浆葡萄糖水平。糖原异生受p53介导的PGC1α/β及其下游效应物葡萄糖-6磷酸葡萄糖(glucose-6 phosphate, GLC-6-P)和磷酸烯醇丙酮酸羧激酶(phosphoenolpyruvate carboxykinase, PEPCK)的调控。mTERT或PGC1α的表达增强或p53的遗传敲除可提高PGC1α/β、GLC-6-P和PEPCK的表达,恢复糖原异生水平。端粒功能障碍引起的线粒体损伤则可以增强组织对葡萄糖代谢的依赖。糖酵解与线粒体氧化代谢比值的增加也可能导致NAD/NADH的变化,进一步降低SIRT活性。另外,端粒功能障碍引起的DNA损伤和PARP激活会减少NAD,并削弱SIRT功能。聚腺苷二磷酸核糖聚合酶(Poly (ADP-ribose)polymerase, PARP)和SIRT竞争NAD+,因此PARP1基因敲除小鼠中可以观察到抑制PARP会导致棕色脂肪组织和肌肉中SIRT功能活性的增加。
炎症
衰老过程会涉及来自基因组受损的细胞或衰老细胞的炎症信号。端粒功能障碍可以在多个水平引发和维持炎症。首先,端粒功能异常会引起细胞衰老,从而刺激IL-6和TNF-α和其他炎症因子的产生和分泌。其次,端粒功能障碍会导致染色体外片段的产生,这些片段可通过激活环GMP-AMP合酶(cyclic GMP-AMP synthase, cGAS)/干扰素基因刺激物(stimulator of interferon genes, STING)途径来促进细胞自噬死亡。确实如此,最近的一项癌症相关研究表明,用新药6-thio-2’-deoxyguanosine(6-thio-dG)靶向端粒会诱导TIF的产生以及染色体外DNA向肿瘤微环境中的释放。树突状细胞摄取此DNA会触发细胞内cGAS/STING途径,并通过干扰素调节因子3(interferon regulatory factor-3, IRF3)TANK结合激酶1(TANK binding kinase-1, TBK1)介导的I型干扰素上调引发炎症级联反应,从而导致产生干扰素γ(interferon γ, IFNγ)的CD8+ T细胞交叉激活和富集。最近的研究表明端粒功能障碍和组织炎症之间有直接的联系(图5)。具体来说,端粒功能异常会激活共济失调的毛细血管扩张突变(ataxia telangiectasia mutated, ATM)/Abelson鼠白血病病毒癌基因同源物1 (Abelson murine leukemia viral oncogene homolog 1, cABL)诱导的YAP1磷酸化,导致其向细胞核的转移。定位细胞核中的YAP1可以上调炎症和炎症小体基因表达,如pro-IL18,NLRC5,NLRP1b和NLRP6。脂酶激活的caspase-1与肠道微生物组激活的炎症小体轴一起,将pro-IL-18剪切成IL-18,IL-18进而招募并激活T细胞从而引发炎症。用YAP1抑制剂、caspase-1抑制剂或抗生素对端粒功能障碍小鼠进行体内治疗,或在肠腔室中重新激活端粒酶减少procaspase-1向caspase-1的剪切,以及成熟的IL-18产生,可以缓解组织炎症。Chakravarti等将端粒功能障碍和炎症反应之间建立了直接联系,部分解释了在老年人群中出现的与年龄相关的炎症反应的增加。该研究还强调了细胞内端粒功能障碍驱动的分子途径与微生物组在驱动炎症反应中的合作。
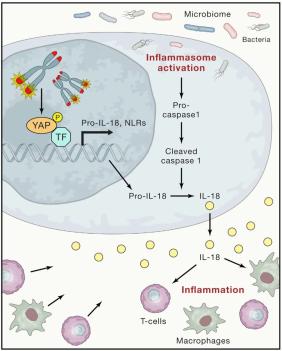
图5 端粒功能障碍导致组织炎症
端粒功能障碍通过激活ATM/cABL/YAP1轴并驱动成熟IL18分泌以募集并增强T细胞和巨噬细胞而驱动组织炎症。ATM,共济失调毛细血管扩张突变;cABL,Abelson鼠白血病病毒致癌基因同源物1;YAP1,相关蛋白1;IL-18,白介素18。
总而言之,许多研究将端粒动力学与衰老的所有特征相关的途径和生物学过程联系在一起。此外,这些压力反应中许多都会产生反馈回路,从而进一步损害端粒,放大并加速退行性衰老。端粒几乎与衰老的所有特征都有内在的相互联系,这导致了衰老的开始与加重。
端粒和端粒酶在年龄相关疾病和癌症中的作用
通过对端粒维持基因编辑小鼠和个体的研究,揭示了端粒是如何促进衰老以及诱发衰老相关的疾病的。小鼠和人类端粒存在长度和调控机制水平上存在差异。鼠(Mus musculus)的端粒长度是人类端粒的10倍多(小鼠30-150 kb,人类10-15 kb)。相对于人类体细胞中的端粒酶,小鼠体细胞中端粒酶活性更高。这些物种的差异暗示了端粒和端粒酶在基因组稳定性,衰老和癌症中的功能,促使人们通过工程改造产生具有较短的类人端粒的小鼠来阐明这些功能和机理。在缺失TERT或TERC的小鼠中,连续的代际杂交逐渐缩短了端粒,最终在第3代时出现了端粒功能障碍(末段-末段融合)。这种端粒功能障碍的表现与广泛的组织干细胞耗竭一致,如进行性组织萎缩,生殖细胞耗竭,生殖力降低,适应性免疫力降低,记忆力下降,伤口愈合速度延迟,应激反应减弱,头发变白和脱发增加,心脏功能减弱,骨质疏松,癌症发病率增加,机体虚弱。
本节总结了很多端粒功能与衰老过程有关的证据,如衰弱以及与衰老相关疾病,包括心血管疾病(动脉粥样硬化,血管性痴呆和冠状动脉疾病),代谢性疾病(II型糖尿病),神经系统疾病(帕金森氏病)和癌症。
衰老相关疾病中的端粒功能障碍和端粒酶
大量研究表明,端粒功能障碍经常出现在衰老相关疾病中。首先,在皮肤,胃肠道和造血系统等细胞大量增殖的组织中,祖细胞隔室中端粒酶水平降低和组织不断更新会导致渐进性端粒磨损,最终触发DNA损伤反应,例如细胞周期停滞,细胞凋亡,分化受损和衰老。其次,心脏,大脑和肝脏等细胞增殖较少的组织可能会经历ROS诱导的端粒序列损伤,并随着时间的流逝引起端粒序列受损和脱帽。具体而言,鼠和人体研究表明,氧化应激本身会加速血管内皮、骨骼肌、心肌细胞以及先天免疫和适应性免疫的相关免疫细胞的端粒磨损。此外,尽管端粒缩短本身会产生TIF,但ROS诱导的鸟嘌呤修饰也会产生TIF,说明了由于端粒蛋白复合体脱离是导致端粒脱帽的根本原因。从开创性研究中可以明显看出这一点,防护蛋白成分的干扰(例如TRF2的显性负突变形式)可以诱导TIF,而不影响端粒长度。
衰老和炎症的纠缠过程可能与端粒衰老有着特别的联系。这些多效行为可诱发动脉粥样硬化,II型糖尿病,骨关节炎以及帕金森氏症和阿尔茨海默氏病。最近的一项研究强调了衰老细胞在诱发阿尔茨海默氏病中的作用。在这项研究中,产生了神经原纤维缠结和类似阿兹海默氏症表型的Tau突变(MAPTP301SPS19)的小鼠,在通过遗传或药物方法去除表达p16INK4a的星形胶质细胞和小胶质细胞后,小鼠认知功能有所保留。这项研究发现了衰老细胞的积累先于神经原纤维缠结的形成,这表明衰老细胞可能诱导缠结的形成。相似地,从BubR1早衰小鼠模型中去除衰老细胞会延长健康寿命。这些研究促进了能够清除人类衰老细胞的senolytic药物的研发。衰老-炎症性反应轴在患有晚期心力衰竭,心脏肥大和冠状动脉疾病的患者中也可能激活。
炎性疾病中的端粒功能障碍
端粒病的研究表明,端粒功能障碍可导致人类衰老中的炎性疾病。端粒病变是由端粒维持基因,包括TERT,TERC,DKC,PARN,RTEL1,TINF2和POT1的种系缺陷造成的。在细胞水平上,端粒病有以下特征:(1)造血干细胞耗竭导致骨髓衰竭;(2)淋巴细胞免疫衰老;(3)肠道干细胞损失导致与绒毛细胞凋亡相关的肠道绒毛萎缩,绒毛变钝,基底浆细胞增多和上皮内淋巴细胞增多。在组织水平上,端粒病显示出特发性肺纤维化,肝硬化和肾脏疾病的发生率增加,所有这些都与炎症加剧有关。鉴于高ROS水平与端粒缩短有关,并且被认为是组织炎症的驱动力,因此即使在端粒维持基因没有突变的情况下,端粒缩短和损伤也可能诱发和助长老年人的各种炎性疾病。这些情况可能源于受影响组织的一些细胞中出现的端粒功能失调,导致炎症细胞因子局部增加,并进一步导致组织损伤和端粒缩短。这类炎性疾病包括炎症性肠病,胰腺炎,非酒精性脂肪肝,慢性阻塞性肺疾病,以及慢性肝病导致的肝硬化等。在这种情况下,端粒功能障碍可能会使致病因素的致病作用更强,且端粒功能障碍自身也作为一种致病因素使疾病的发生和发展更为迅猛。
线粒体功能障碍及ROS升高都与衰老有关,特别是与衰老和炎症有关。这些疾病治疗的主要目标是维持线粒体活性,因为对鼠类的研究表明PGC1α过表达对各种与衰老相关的生物过程(包括代谢和ROS)具有有益作用。已知不受控制的ROS会影响糖异生,脂肪酸代谢和β-氧化,这可能引发与衰老相关代谢性疾病,包括糖尿病、癌症、炎症反应、肌肉减少症、神经退行性疾病(例如阿尔茨海默氏病,帕金森氏病,多发性硬化症和肌萎缩性侧索硬化症)以及个体变得更加虚弱。
端粒功能障碍,端粒酶和癌症
癌症是一种衰老相关疾病,在美国80岁老人中每2名男性和每3名女性中就有1人患有癌症。此外,上皮癌是老年人的主要癌症类型。这些癌症细胞通常是非整倍体,并具有许多染色体结构改变。然而随着小鼠年龄的增长,出现最多的癌症类型主要是造血(淋巴瘤)和间充质(肉瘤)癌,很少有上皮癌。而且,这些鼠类恶性肿瘤很少有染色体结构畸变。这些跨物种的差异可能阐明驱动老年人上皮癌占优势的关键机制,以及这些癌症中基因组不稳定的基础。我们通过对拥有更短的类人端粒的端粒酶缺陷小鼠进行基因编辑,证明了在p53缺失的情况下,端粒功能障碍能够使处于危机中的细胞在染色体断裂事件中存活下来,从而促进癌症相关基因的扩增、缺失和易位基因编辑,与人类相似(图6)。令人惊讶的是,这些TERC/p53-null小鼠不仅癌症发病率增加,还显示出具有复杂核型的人源化肿瘤图谱。这些小鼠实验以及随后的早期人类癌症中端粒功能异常的基因组证据确定了端粒功能障碍是诱发老年人上皮癌变的主要机制。换句话说,小鼠中这种突变机制的缺乏(长端粒和端粒酶的混杂表达)可以保护小鼠免受上皮癌的侵害。
上述小鼠模型揭示了端粒在癌症演变中的复杂作用,尤其是端粒功能障碍如何在限制完全恶性进展的同时促发肿瘤。例如,在腺瘤性大肠菌病(APCmin)的肠腺瘤中,TERC-null小鼠模型中的端粒功能障碍在一开始会提高腺瘤的发生率,但最终会限制肿瘤发展为大腺瘤并提高生存率。这种受限制的恶性进展主要与存在端粒功能障碍的恶性肿瘤细胞中p53激活有关。p53肿瘤抑制因子的失活是人类上皮癌中最常见的事件之一,p53的状态可以决定端粒功能障碍是促进还是抑制了癌症的发展。特别是在缺乏p53的晚代TERC-/-小鼠上表现出癌症发病率增加和存活率降低,而缺乏Ink4a/Arf的晚代TERC-/-小鼠上表现出明显的癌症发病率降低,存活率升高。值得注意的是,即使缺乏Ink4a/Arf的小鼠缺乏p19ARF(在致癌激活过程中发出p53信号),这些小鼠中仍保留了功能性p53依赖的DNA损伤反应(例如凋亡)。因此,端粒在肿瘤发生中的作用取决于p53介导的DNA损伤信号传导和相关细胞检查点反应的完整性。
对人类癌症的最新单细胞DNA测序发现,许多癌症是由于单一的不定时基因组突变演化而来,其中拷贝数改变和突变的爆发在所有测序结果中占主要地位。尽管关于驱动这种间歇性进化或“发作性不稳定性”的机制正在积极被研究,但小鼠和人类的证据清楚地表明基于端粒的危机和随后的端粒酶重新激活是形成癌症基因组并诱发老年人上皮癌变的机制。虽然这种突发性的不稳定性模型与人类基因组学数据一致,但我们认为“Vogelstein机制”— 驱动癌变突变的逐步累积—也可能在这里起作用,并且两个模型不是互斥的。
其他支持端粒-癌症联系的证据包括老年个体结肠粘膜中出现年龄依赖性端粒缩短和癌症组织中的端粒比邻近的非恶性组织短。此外,与相应的正常组织相比,人类肿瘤组织中TERT和TERC的表达升高。人类原代细胞的恶性转化需要TERT增强表达,这表明端粒的维持对于完全恶性转化至关重要。最后,用诱导型TERT等位基因改造的易癌小鼠体内首先会经历端粒危机,随后才是端粒酶的重新激活。这种危机-再活化序列产生的更有侵略性的肿瘤证明了危机诱发的基因组事件更易导致具有侵略性的恶性表型发生。类似地,在端粒酶野生型小鼠中瞬时诱导端粒解封(通过突变体TRF2蛋白破坏和庇护素复合物破坏)会增加在用致癌物治疗的小鼠中肝细胞癌的发生和发展。相反,在致癌物诱导的肝细胞癌模型中,TERC基因敲除的小鼠模型中的端粒缩短会增加肿瘤的发生概率,但不能促进肿瘤发展。综上,多种证据分别证实了端粒危机和端粒酶再活化在癌症发生和进展中的作用。
端粒和端粒酶依赖的致癌机制
如前所述,端粒功能的丧失会导致末段-末段融合。在有丝分裂过程中,这些双着丝粒染色体会在后期连接,最终使染色体断裂,从而导致出现由Creighton和McClintock在1931年首次提出的BFB循环。幸存的BFB子细胞在危机期间会积聚与癌症相关的染色体变化,并最终激活端粒酶,抑制DNA损伤信号传导并缓解严重的染色体不稳定状态,进而导致恶性肿瘤的全面进展。一个TERT诱导型前列腺癌模型已通过实验验证了癌症基因组进化概念。在这种癌症模型中,与端粒相对完整的对照组相比,癌细胞中端粒严重缩短,会导致凋亡增加和增殖减少,从而产生体积较小且侵袭性较小的肿瘤。在这些染色体不稳定的高级前列腺上皮内瘤变细胞中,端粒酶的实验性激活会导致肿瘤的恶性进展,包括产生骨转移的新表型。相比之下,端粒完整的对照组中仅表现出局部侵袭。此外,对经历端粒危机的肿瘤进行的基因组分析显示了额外的转移前突变(例如,SMAD4缺失和对转化生长因子β(transforming growth factor-β, TGF-β)途径的额外干扰)。因此,端粒危机驱动染色体的不稳定性会引发癌症,随后的端粒酶重新激活使恶性肿瘤全面进展。
大规模测序研究表明,TERT启动子突变是人类癌症中最常见的非编码突变。此类突变在自身更新率较低的组织癌症中高度富集。例如,黑色素瘤显示出高度复发的TERT启动子突变,平均TERT表达增加4倍;原发性胶质母细胞瘤在80%以上的病例中显示TERT启动子突变。TERT启动子中最常见的复发性单核苷酸突变发生在G228A和G250A,它们产生从头ETS共有结合基序来募集GA结合蛋白和潜在的其他E26转化特异性转录因子家族以提高TERT表达。导致癌症中TERT表达增加的其他机制也是由危机诱发的染色体改变所致,这会在肝细胞癌中产生TERT基因座的局灶扩增。最后,基因治疗研究表明,CRISPR介导的将TERT启动子序列矫正为野生型序列显著延长了神经胶质瘤小鼠的存活时间。
除了通过基因组机制驱动TERT的上调外,致癌信号通路还可以增强TERT基因的转录。细胞MYC(c-MYC)可以结合TERT启动子中的Myc结合元件来诱导TERT转录并增加人类原代成纤维细胞的端粒酶活性,尽管仅增强TERT的表达不足以替代c-MYC促进转化作用。相似地,激活的WNT信号传导能与Krüppel样因子4(Krüppel-like factor 4, KLF4)协同激活TERT转录。虽然发育和癌症相关的关键通路可以激活TERT,但也有证据表明TERT可以顺次激活这些途径。TERT过表达小鼠模型中端粒酶的激活表明,静止的毛囊干细胞和肾脏足细胞可独立于端粒长度、TERC功能或TERT逆转录酶功能而发生增殖。研究表明,TERT直接结合TCF元件并增强MYC和WNT转录程序,这是已知的能够稳定干细胞和驱动癌症的途径。另外,TERT与NF-κB复合物相互作用并结合IL-6和TNF-α启动子元件,增加细胞增殖和对死亡的抵抗力。抑制p65可以减弱异位TERT和TERC表达后增加的细胞增殖能力和对细胞死亡的抵抗力。从功能上讲,G1 TERC-null小鼠对内毒素休克的抵抗力要高于对照,超过50%的小鼠存活于脂多糖诱导的休克,而对照组则为25%。这表明端粒酶调节NF-κB的炎症反应独立于端粒长度。这些研究指向致癌信号分子和TERT在癌症相关回路调节中的相互作用。需要进一步的研究来评估这些通路在人类癌症中的作用。
尽管尚未确定遮蔽复合物在癌症发生发展中的确切作用,但已在癌症中检测到了这些蛋白发生突变和/或表达改变。在早期慢性淋巴细胞白血病,端粒功能障碍与疾病的发生和端到端融合有关。在这一疾病中遮蔽复合物中蛋白的表达降低,如TRF1,RAP1和POT1以及TIN2和TPP1。在这些蛋白里,POT1是癌症最易发生突变的蛋白。慢性淋巴细胞性白血病病例中,其中体细胞突变占5%。POT1或RAP1的体细胞突变可发生在家族性神经胶质瘤、家族性黑色素瘤、Li-Fraumeni样综合征、甲状旁腺腺瘤、心脏血管肉瘤、套细胞淋巴瘤和肝癌中。TRF1和TRF2也在多种肿瘤中上调,包括肺癌、乳腺癌、结肠癌、胃癌和肾癌,暗示着遮蔽复合物在人类癌症中的支持作用。
端粒维持在癌症中的替代机制
尽管通过端粒酶维持端粒是癌症中最常见的端粒维持机制,但癌细胞还是利用了不依赖端粒酶的重组介导的机制,称为端粒替代性延长(alternative lengthening of telomere, ALT)。ALT发生在5%–15%的人类癌症中,尤其是在骨肉瘤和胶质细胞瘤中,并且往往与预后不良相关。ALT使用DNA同源重组的修复机制延长端粒,其中端粒DNA模板从姐妹染色单体或非同源染色体复制而来。ALT机制的重要因子包括MRE11,RAD50,NBS1,FEN1,MUS81和FANCD2。有趣的是,ALT阳性细胞通常在感知胞质DNA的能力上存在缺陷,并且染色体外端粒重复(extrachromosomal telomere repeat, ECTR)通过cGAS-STING胞质DNA传感途径诱发IFN反应。因此,由于ALT的不断产生,采用ALT的癌细胞需要克服其他抗增殖障碍,例如衰老诱导和先天免疫监视。有趣的是,尽管ALT是在没有端粒酶重新激活的情况下诱发肿瘤的重要因素,但在驱动肿瘤侵袭性恶变和转移方面效率较低。多次传代的小鼠mTerc-/- Ink4a/Arf-/-成纤维细胞中出现ALT的发展。尽管ALT阳性细胞的端粒得以维持,但它们无法形成肺转移。然而在mTERC转导后,端粒酶阳性的癌细胞具有了转移潜能,这证实表现出ALT阳性和端粒酶阳性的癌症在生物学上并不等效。
ALT途径还可以用作端粒酶抑制剂治疗的耐药机制。在用他莫昔芬控制的TERT等位基因(TERT-ER)编辑生成的ATM突变小鼠(淋巴瘤易感)模型中,端粒酶活性的消失导致淋巴瘤细胞重新进入端粒危机。尽管端粒酶的消失治愈了三分之二的小鼠,但其余的小鼠中表现出有ALT标志性特征的复发性肿瘤。有趣的是,这些复发性肿瘤在已知的涉及同源重组的基因(在ALT以及PGC1β位点发挥重要作用的基因)中获得了扩增和缺失,这可能表明ALT阳性细胞中存在持续的线粒体应激。这些机制提供了将端粒酶抑制剂与靶向同源重组或线粒体机制的药物相结合的方案。对于后者来说,表现出线粒体功能减弱和相关的胞内高ROS水平的ALT阳性癌细胞特别容易受到靶向抗氧化蛋白质的影响。
端粒酶治疗的前景与未来展望
端粒功能障碍与衰老标志物、衰老相关疾病的发生以及遗传和获得性退行性疾病的发展的联系,促使人们将端粒酶修复作为一种潜在的延缓衰老策略(图6)。利用这种思路进行治疗的最佳方式可能是瞬时端粒酶诱导,即通过它恢复端粒储备和修复端粒损伤,同时避免由组成型端粒酶激活而促进癌症的可能性。在具有长端粒的临床前小鼠模型中,TERT表达将小鼠寿命延长了40%。尽管这些小鼠还携带了p53、p16和ARF的额外拷贝(增强了小鼠对癌症的抗性)。尚待确定的是,延长的TERT表达来延长寿命和缓解疾病是否与TERT对端粒的作用或WNT的活化有关,其中WNT的活化可能增强干细胞的储备
随着对端粒酶调节的分子网络的以及激活端粒酶促进早衰小鼠年轻化的进一步了解,人们逐渐关注可以激活TERT表达的抗衰老药物。尽管对它们的作用机理还不甚了解,但已经有几种小分子,包括TA-65(环黄芪醇)和组蛋白脱乙酰酶抑制剂,被证明可以激活TERT(图6)。TA-65是从黄芪属植物中分离出来的一种天然化合物。正在进行的人体试验表明,TA-65改善了黄斑功能,降低了高密度脂蛋白水平,并降低了炎症标志物c-反应蛋白和TNF-α水平。此外,激素类药物,如对患有端粒病的个体使用达那唑(一种抗雌激素和抗孕激素)和5a-二氢睾丸激素(一种雄激素)可以增加端粒酶水平。最后,靶向TERC稳定性的药物可能为治疗端粒病提供新的选择。PAPD5通过3’寡腺苷酸化来降解TERC,然后诱发RNA外泌体破坏这些转录本。如前所述,与TERC的去腺苷酸化和成熟有关的PARN在包括先天性角化不全在内的几种疾病中发生了突变。一种PAPD5小分子抑制剂可增加来源先天性角化不全患者的诱导多能干细胞的端粒长度,并且可在小鼠中可长期发挥作用。
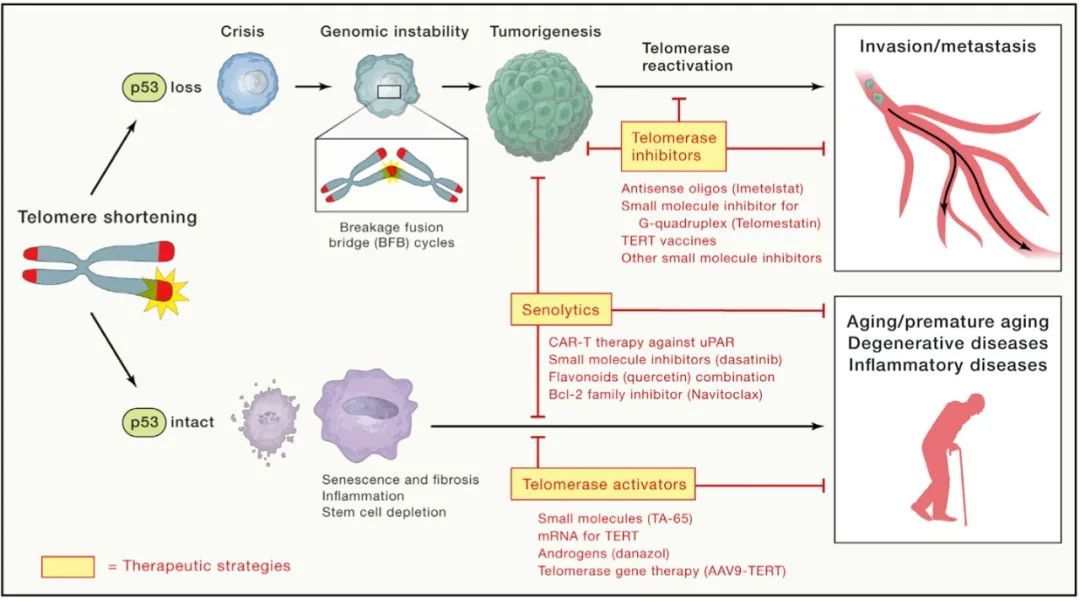
图6 衰老,癌症和潜在疗法中的端粒/端粒酶
存在功能性p53检查点时,端粒缩短会导致衰老,纤维化,炎症和干细胞耗竭。这些过程导致衰老以及其他退行性和炎性疾病。端粒酶激活剂和senolytics可以抑制这些过程,并抑制衰老和与年龄相关疾病。端粒缩短也会导致端粒融合和断裂-融合-桥循环。在没有p53检查点的情况下,这些事件会导致肿瘤发生。端粒酶的进一步激活导致肿瘤侵袭和转移。端粒酶抑制剂和senolytics抑制可阻碍肿瘤进展,侵袭和转移的过程。
脉冲式端粒酶活化疗法的一种特别引人入胜的应用可能是对诸如Werner和Bloom综合征等早衰性疾病的治疗。这一方法源于人类遗传退行性疾病相关的基因缺失小鼠出现退行性表型极度缺乏的现象。包括(1)由ATM的突变失活导致的共济失调-毛细血管扩张;(2)Bloom综合征,由RecQ解旋酶家族的BLM突变引起;(3)杜氏肌营养不良症(Duchenne muscular dystrophy, DMD),由肌营养不良蛋白的突变引起。令人惊讶的是,当这些等位基因中的每一个分别与TERC基因敲除小鼠杂交时,随着端粒变得更短,更像端粒在人体内的长度(通常在G2-G3时),这些综合症的表型渐渐出现。例如,WRN/TERC-null小鼠概括了Werner综合征的标志性特征,包括后凸畸形、病理性长骨骨折、脱发和头发变白、干细胞和免疫区室缺陷、老年性白内障、造血功能障碍(例如全血细胞减少和对反应的不平衡)免疫原和代谢缺陷,包括胰岛素抗性。
如上所述,在WRN或BLM基因中发生突变的端粒完整小鼠模型没有表现出任何退行性表型,这为端粒酶激活疗法提供了理论依据,以延迟或减轻症状并延长预期寿命。与此类似,在生殖系突变影响端粒维持的基因的个体中(例如DKC),端粒酶激活疗法可以缓解进行性症状,例如贫血,肺纤维化和胃肠道功能障碍。端粒酶激活可能起有益作用的另一类治疗选择有限的疾病是慢性炎性疾病,例如肝硬化,胰腺炎和溃疡性结肠炎(图6)。疾病发端的端粒功能障碍可诱发组织炎症,继而可加速端粒缩短,形成正反馈循环,最终导致疾病复发,甚至诱发基因组不稳定,p53丢失和端粒酶重新激活所致的癌症。在进入端粒危机之前的疾病的非常早期阶段,端粒酶的激活可以预防疾病发作和癌症发生。鉴于在端粒酶的基因诱导后小鼠脑部得到了显着改善,端粒酶激活也可能用于治疗神经退行性疾病。除端粒酶激活外,还可能通过削弱介导功能障碍性端粒的组织破坏作用的检查点的功能,来改善端粒功能障碍引起的器官变性和死亡。沿着这些思路,已经证明选择性抑制p21依赖的细胞周期阻滞或puma介导的凋亡可以改善端粒功能失调小鼠组织完整性、延长寿命,而不促进癌症的发展。Exo1的缺失也可以通过抑制染色体融合体和BFB循环的形成来增强端粒功能障碍小鼠对DNA损伤的感应和p53检查点的激活,从而使端粒功能障碍小鼠的组织保持完整和稳定,并延长小鼠的寿命。
与端粒酶激活在延缓衰老治疗中的潜在应用相反,在大多数癌症中观察到的端粒酶活性增加促进了人们开发抗端粒酶治疗剂,包括反义寡核苷酸,疫苗和小分子抑制剂等几种策略已经被设计出以针对癌症中的TERT,但是还没有抗端粒酶药物进入III期临床试验。这种效率有限性可能要归因于,端粒缩短至可抑制肿瘤的长度所需的时间。此外,抑制端粒酶的替代策略可能在临床上产生更有意义的效果。首先,由于其功能性检查点机制会触发衰老,具有完整p53的癌症可能更适合端粒酶抑制。临床前动物研究表明,TERT抑制可导致淋巴瘤中ALT途径的激活,因此该策略仍需谨慎选择。端粒酶和ALT通路抑制药物的联合使用可以最大程度地减少耐药性的出现。在ALT阳性的癌症亚群中,靶向相关的免疫回路也可以提升药物反应率和预后效果。也就是说,因为ALT阳性癌细胞中胞质DNA能持续产生,而胞质DNA通过激活cGAS-STING通路来上调IFN信号,人们很容易推测出这些肿瘤可能对免疫疗法有更高的反应。在临床前研究中,将核苷类似物6-thio-dG掺入新合成的端粒中导致端粒DNA损伤,可使对PD-L1耐药的癌症对免疫检查点疗法敏感。综上所述,尽管确定基因型和生物学背景以及与协同作用的特定联合治疗方法非常重要,抗端粒酶疗法仍然具有可行性,因为它在抑制晚期恶性肿瘤方面具有潜在的作用。
最后,目前对染色体结构和细胞生物学基础知识的探索阐明了多种人类疾病和衰老本身的核心机制。端粒领域已成为基础科学和多学科融合的典范。虽然端粒和端粒酶在衰老和癌症发病机制中的很多作用已经被揭示,但许多知识空白依然存在,例如调控端粒酶表达和活性的机制,TERT的非经典功能以及端粒功能障碍与病理过程(如炎症,纤维化和退行性疾病)之间的相互作用。不论端粒损伤是人类疾病的诱因还是仅仅是影响因素,它都在人类疾病的发生发展过程中起到了“不可或缺”的作用。这种主要作用促进了人们去开发和测试端粒酶激活剂来治疗衰老和与衰老相关疾病,并评估用于治疗晚期癌症的有效的端粒酶抑制剂。本篇关于端粒的综述希望激发人们进一步研究端粒和端粒酶的作用,这可能有助于应对我们所有人最终都要遭受的致命疾病——衰老。

来源:老顽童说
原标题:《【重磅综述】从过去到未来—解密端粒与衰老》